
Abstract
Obese pregnant women may transmit their metabolic phenotype to offspring, leading to a cycle of obesity and diabetes over generations. Early childhood obesity predicts nonalcoholic fatty liver disease (NAFLD), the most common chronic human liver disease. The fetus may be vulnerable to steatosis because immature fetal adipose depots are not available to buffer the excess transplacental lipid delivery in maternal obesity. In animal models, in utero high-fat diet exposure results in an increase in the accumulation of liver triglycerides in offspring and increased hepatic oxidative stress and apoptosis, perhaps priming the liver for later development of NAFLD. Innate immune dysfunction and necroinflammatory changes have been observed in postnatal offspring liver of animals born to high-fat–fed dams. Postweaning, livers of offspring exposed to maternal high-fat feeding in utero share pathophysiologic features with human NAFLD, including increased de novo lipogenesis and decreased free fatty acid oxidation. Human studies using magnetic resonance imaging have shown that maternal BMI predicts infant intrahepatocellular lipid storage, as seen in animal models. The generational transfer of NAFLD may occur via epigenetic changes in offspring liver. Transmission of microbiota from mother to infant may impact energy retention and immune function that contribute to a predisposition to NAFLD.
Similar content being viewed by others


Main
Under normal metabolic conditions, hepatic fat should not exceed 5% of total liver weight. The liver is a through station for dietary lipid and fatty acids (FA) liberated from adipose tissue. The liver then repackages lipid as lipoprotein or generates metabolic intermediates that can then be trafficked to tissues in need of energy. In obesity, metabolic conditions favor the storage of fat in the liver. The spectrum of disease due to hepatic fat storage is termed nonalcoholic fatty liver disease (NAFLD). Within hepatocytes, lipid can be partitioned away from normal cellular activity with relatively little clinical consequence, leading to steatosis. Hepatic fat storage can also drive the inflammatory response nonalcoholic steatohepatitis. Chronic hepatic inflammation can then lead to fibrosis, cirrhosis, and end-stage liver disease in some individuals. With the rise of obesity, it is predicted that NAFLD will be the most common etiology for liver transplantation in the 21st century (1).
Prevalence estimates of NAFLD in children range from 3–10% in Western societies, approaching 70% in obese children (2). NAFLD diagnoses increase with age in childhood, and its prevalence in adolescents has more than doubled in the last two decades (3). Like adults, children with NAFLD can progress to cirrhosis and end-stage liver disease (4). NAFLD is more common in boys than girls, and there are distinct differences between race/ethnic groups in NAFLD prevalence (5). Compared to Caucasian and Hispanic adults, African Americans are protected from the accumulation of hepatic fat even when BMI and insulin sensitivity are controlled for (6). The diagnosis of NAFLD is also strongly predictive of other features of metabolic disease, including insulin resistance, hypertension, dyslipidemia, carotid atherosclerosis, and intramyocellular lipid deposition (7,8,9,10). A unique feature of pediatric NAFLD is the predilection for children to deposit fat and develop inflammation in the periportal region vs. the more classic perivenular distribution seen in adults (11). This difference is not well understood but is clinically relevant because periportal inflammation is associated with advanced liver disease (12).
The earliest origins of NAFLD and other metabolic diseases may lie in early life, even in utero, experiences. The transmission of metabolic phenotypes from mother to offspring may lead to a generational transfer of risk for a multitude of adverse metabolic outcomes, including NAFLD. In this review, we present data from clinical and basic science research implicating increased fuel exposure in fetal life in the pathogenesis of offspring NAFLD.
Maternal Obesity and The Human Fetus
The prevalence of prepregnancy maternal obesity and gestational diabetes mellitus has climbed steadily over the last half-century (13,14). As many as 60% of women are overweight at the time of conception and, in low-income communities, ~30% of pregnancies in the United States begin with a maternal BMI greater than 30 kg/m2 (15). The epidemiologic association between maternal weight and early childhood obesity has been well described (16,17,18). The recent prospective study by Stamnes Køpp et al. (16), which followed a large cohort of mother/infant pairs and controlled for factors such as paternal weight, maternal exercise, duration of breastfeeding, and household media exposure, found that maternal obesity (BMI > 30) conferred a 0.5 unit increase to offspring BMI at 3 y of age. Evidence from studies in the Pima Indians also implicates exposure to gestational diabetes mellitus as a major risk factor for offspring development of type 2 diabetes in adolescence/early adulthood (19).
Increased hepatic fat storage in offspring of obese mothers may be driven, in part, by increased delivery of nutrients to the fetus. It is neither ethically nor practically possible to directly measure placental nutrient transfer in early pregnancy in humans, but there are several indirect lines of evidence in humans that support the idea that maternal obesity leads to increased fuel availability for the fetus. Studies correlating maternal glucose and lipid excursions with fetal outcomes have consistently shown an impact on fetal growth. The Hyperglycemia and Adverse Pregnancy Outcome study demonstrated increased birth weight associated with maternal glucose levels below those conventionally used to diagnose gestational diabetes (20). Harmon et al. utilized continuous glucose monitoring in pregnant women to demonstrate that significant daytime glucose excursions in obese but nondiabetic mothers correlated with infant adiposity. This study also showed the association of elevated maternal triglycerides and fatty acids with infant adiposity (21). Increased triglyceride content in the placentas of diabetic pregnancies, compared to nondiabetic pregnancies, is also suggestive of higher lipid flux (22).
Increased placental transfer of maternal fuels such as glucose and FA may present a particular difficulty to the fetus early in pregnancy, because subcutaneous fat is not available as a storage buffer to handle excess nutrition. Fetal subcutaneous adipogenesis progresses throughout gestation, but lipid storage remains dormant until the third trimester, when fetal fat accretion accelerates dramatically until parturition (23). This storage process is timed to the development of maximal physiologic insulin resistance in the pregnant mother, allowing for increased glucose and lipid to reach the placenta and ultimately the growing fetus (24). This transfer of maternal fuel to the fetus in the third trimester represents a considerable metabolic investment by the mother. Evolutionarily, this maternal investment may have contributed to increased survival chances for offspring (25). However, the fact that subcutaneous lipid storage is a relatively late event in fetal life may handicap the fetus in its ability to adapt to prepregnancy maternal obesity and insulin resistance. In considering the physiologic challenge of metabolic regulation without robust adipose tissue availability for fat storage, a reasonable comparison can be made to humans with congenital generalized lipodystrophy, an autosomal recessive disorder characterized by the inability to store fat in adipose tissue. Patients with lipodystrophy utilize organs such as liver, pancreas, and skeletal muscle for fat storage, leading to severe insulin resistance and NAFLD despite a normal BMI (26). We hypothesize that in the setting of maternal obesity and gestational diabetes mellitus, the fetus similarly utilizes organs such as liver for energy storage.
Recent human studies have supported this theory. The development of advanced imaging techniques using magnetic resonance technology has allowed for precise measurement of neonatal hepatic fat using methods that are noninvasive and do not expose the research subject to ionizing radiation. Using magnetic resonance spectroscopy, along with others, we have shown that maternal obesity and gestational diabetes mellitus predict neonatal hepatic fat storage (27,28). Across the entire cohort of both normal weight and obese mothers, we found a statistically significant correlation between maternal prepregnancy BMI and offspring intrahepatocellular lipid. However, mother’s gestational weight gain and the infant’s postnatal weight gain did not correlate with infant intrahepatocellular lipid. Importantly, increased fetal hepatic fat storage was independent of overall adiposity in the newborn, suggesting that drivers of hepatic fat storage and subcutaneous adipose fat storage may be different during fetal life.
Fetal exposure to gestational overnutrition leads to increased expression of proinflammatory cytokines in the hypothalamus as well as apparent alterations in hypothalamic sensitivity to key hormonal regulators of appetite and metabolism (29,30). These changes could then be postulated to impact postnatal appetite and weight gain later in life. However, the effect of altered appetite regulation may be limited in the first year of life in humans, when the offspring of obese mothers are more likely to experience “catch-down” growth (31). Male nonhuman primates (NHP) exposed to maternal high-fat diet (HFD) have reduced sympathetic innervation of the liver, an interesting finding in light of research showing that hypothalamic inflammation can impact pancreatic islet cell function via its sympathetic innervation (32,33). However, it is still unclear what effect perturbations in the developing nervous system might have on hepatic metabolism.
One challenge in the study of the impact of maternal obesity on fetal fat storage is the selection of a proper animal model. The term human infant is composed of 12–15% body fat after a healthy pregnancy (34). With the exception of the guinea pig, laboratory animals are born with considerably less subcutaneous fat and without this buffer may be more susceptible than humans to adverse effects of excess fuel transfer from mothers. Nevertheless, studies in a variety of animal models have provided invaluable insight into the mechanisms by which fetal hepatic fat storage may have longstanding metabolic consequences for offspring.
Gestational Overnutrition and Offspring Hepatic Fat Storage
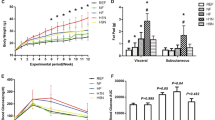
In maternal obesity, the fetoplacental unit develops under conditions of both excess nutrients and inflammation. To study the impact of maternal HFD consumption on the developing fetus, we have used a Japanese macaque NHP model. We have previously demonstrated in our NHP model that consumption of a HFD during pregnancy reduces uterine blood flow, exacerbates placental inflammation, and increases the risk of stillbirth (35). Furthermore, in utero HFD exposure alters the normal development of key metabolic organs in the fetus. Specifically, third trimester fetuses from mothers fed a HFD display steatosis and increased hepatic oxidative stress and apoptosis ( Figure 1 ) (36).

Maternal high-fat diet (HFD) feeding leads to lipid accumulation and activation of oxidative stress in fetal liver. (a and b) Oil Red O staining and (c and d) 4-hydroxynonenal staining (a and c) in fetal liver of controls and (b and d) offspring-HFD (O-HFD) animals. Original magnification, ×20. (e) Fetal liver triglycerides (TGs) were significantly elevated in both O–HFD-resistant (O-HFD R) and O–HFD-sensitive groups (O-HFD S). *P < 0.05 vs. control (one-way analysis of variance with Bonferroni post hoc analysis). (f) As a group, O-HFD animals had a significant increase in the ratio of p-JNK1 to total JNK1 levels. Representative western blots are also shown. *P < 0.05 (two-tailed Student’s t-test). For columns in e and f, black, control: hatched, O-HFD R; and white, O-HFD S. (g) There was a significant correlation between TG levels and the ratio of p-JNK1 to total JNK1 in fetal liver. r2 = 0.27, P = 0.002, n = 6 (O–HFD R); 9 (control and O–HFD S). Reprinted with permission from ref. 36.
In the vast majority of cases, pediatric and adult NAFLD develops in the context of insulin resistance and metabolic syndrome. Limited data in humans have shown that decreases in whole-body insulin sensitivity are present at birth in offspring of obese and insulin-resistant mothers (37,38). However, the NHP model provides evidence that hepatic dysfunction in offspring of obese mothers precedes the development of metabolic syndrome. The steatosis and oxidative stress noted in the liver of the NHP fetus exposed to maternal HFD occurs without an increase in fetal adiposity (36). Thus, fetal steatosis cannot be explained by increased peripheral lipolysis. Indeed, adipose tissue inflammation characteristic of metabolic syndrome is not seen in 1-y-old NHP offspring exposed to maternal HFD (Thorn SR, Newsom SA, Baquero KC et al., unpublished data). Additionally, consistent with our understanding that hepatic de novo lipogenesis (DNL) is marginal in the fetal liver, there was no increase in expression of genes involved in DNL in fetal primate livers exposed to a maternal HFD, suggesting the fetal hepatic phenotype is largely explained by increased transplacental fuel transfer (36). Therefore, it is plausible to conclude that early-life hepatic fat accumulation is not simply an early manifestation of NAFLD but an independent pathophysiologic event that then potentiates postnatal metabolic liver disease. Supportive of this idea are multiple studies demonstrating that exposure to a HFD in utero seems to worsen the NAFLD phenotype in adult mice offspring exposed to a Western-style HFD even postweaning (39,40). In those animals exposed to a maternal HFD, weaning to a standard control diet does not completely reverse NAFLD (39). Moreover, in the NHP, we have found persistent steatosis, even in the absence of obesity, in the juvenile offspring of mothers who develop obesity on a HFD (Thorn SR, Newsom SA, Baquero KC et al., unpublished data).
The postnatal persistence of increased hepatic fat storage may result from changes to any of the following intrinsic hepatic processes: DNL, FA oxidation, or lipoprotein export. Steroid regulatory element–binding protein 1c is an insulin-regulated transcription factor that is principal controller of hepatic DNL. As mentioned previously, DNL is not upregulated in the fetal liver but appears to be primed by exposure to maternal obesity. Steroid regulatory element–binding protein 1c is upregulated in the livers of adult mice and juvenile NHP offspring previously exposed to maternal HFD (39,41) (Thorn SR, Newsom SA, Baquero KC et al., unpublished data). Bruce et al. used two lines of evidence to demonstrate reduced FA oxidation in adult mice that had been exposed to a maternal HFD. When exposed to a postweaning HFD, adult mice exposed to gestational overnutrition were unable to increase mitochondrial β-oxidative gene expression to clear increased levels of intrahepatic lipid. β-Hydroxybutyrate, a ketone body produced via mitochondrial FA oxidation, was decreased in the adult livers of mice exposed to gestational overnutrition compared to controls, suggesting reduced FA oxidation (39). In agreement with the findings of Bruce, Bayol et al. (40) found that hepatic expression of carnitine palmitotyl transferase, a key enzyme that mediates transport of long chain FA into the mitochondria for oxidation, is not increased in the steatotic livers of adult mice who were exposed to gestational overnutrition. Shankar et al. (42) also demonstrated reduction in FA oxidation in offspring exposed to a maternal HFD. Additional studies by his laboratory found reduced expression of the nutrient sensing mitochondrial protein sirtuin 3, a key deacetylase, in the livers of mice born to obese dams, implying that defective nutrient sensing is involved in the mitochondrial dysfunction that results in decreased FA oxidation and steatosis (43).
Another member of the sirtuin family, Sirtuin 1 (SIRT1) is a key chromatin deactelyase that controls a broad range of genes of metabolic importance. SIRT1 regulates transcription of peroxisome proliferator–activated receptor α (PPARα) and peroxisome proliferator–activated receptor γ, which control FA oxidation and lipogenesis, respectively. Lipids and lipid intermediates are the natural ligands for PPARα, resulting in increased PPARα transcriptional activity to promote FA oxidation, but this activation appears to be impaired in patients with NAFLD (44,45). As demonstrated in liver-specific SIRT1 knockout mice, reduced SIRT1 expression and activity decreases PPARα transactivation and increases susceptibility to NAFLD on a HFD (46). In NHP exposed to a HFD, the fetal liver displayed decreased SIRT1 expression and activity that was not seen in fetuses born to obese animals on a healthy diet, suggesting that this change in SIRT1 was driven by maternal HFD exposure rather than maternal obesity (47). Importantly, the fetuses showed increased peroxisome proliferator–activated receptor γ and lower PPARα expression. Finally, exposure to gestational overnutrition leads to an increase in hepatic secretion of very low density lipoprotein in adult mice, even after weaning to a control diet, but this compensatory increase cannot overcome the increased hepatic DNL and decreased FA oxidation seen in these animals and consequently steatosis persists (48).
Gestational Overnutrition and Steatohepatitis
The storage of excess intracellular triglyceride in hepatocytes represents the first hit of the “two hit” theory of the pathogenesis of steatohepatitis. A second hit then likely arises from multiple potential sources, including mitochondrial dysfunction leading to the generation of toxic reactive oxygen species, endoplasmic reticulum stress, bioactivity of lipids such as diacylglycerol or lipid derivatives like ceramide, and systemic inflammatory mediators (49). This second hit then triggers hepatocyte injury, inflammation, activation of hepatic stellate cells, and liver fibrosis (50).
Suggestive of decreased mitochondrial capacity, adult mice from obese dams demonstrate a decrease in hepatic expression of mitochondrial genes involved in the electron transport chain (39). As FAs are mobilized into mitochondria for oxidation, mitochondrial dysfunction may then lead to generation of reactive oxygen species that lead to oxidative stress in the liver (51). Juvenile mice exposed to HFD demonstrate increased hepatic expression of genes coding for antioxidant enzymes, likely a physiologic response to increased reactive oxygen species generation (40). As evidence of cellular stress, fetal livers of NHP exposed to HFD demonstrated increased staining of 4-Hydroxynonenal, a product of lipid peroxidation of cells, as well as increased phosphorlyation of c-jun n-terminal kinase isoform 1 ( Figure 1 ) (36). In monkeys exposed to gestational overnutrition, the juvenile liver showed evidence of increased cellular apoptosis as measured by terminal deoxynucleotidyl transferase nick-end labeling assay. Hepatocyte apoptosis is now thought to play an important role in the progression to steatohepatitis in patients with NAFLD (52). Serum levels of CK-18 fragments, released by apoptotic hepatocytes, have been shown to differentiate steatohepatitis from simple steatosis (53). In a cross-sectional analysis, children suspected of NAFLD had increased CK-18 levels compared to lean and obese controls (54).
In humans, most cases of nonalcoholic steatohepatitis are found in adults, rather than children. Like other chronic inflammatory processes in the liver, progression of nonalcoholic steatohepatitis to clinically relevant hepatic fibrosis takes years if not decades in humans. Most studies using a rodent model of gestational overnutrition have assessed offspring liver outcomes at 3–6 mo, which may be too early to observe steatohepatitis. In contrast, Mouralidarane et al. (55) followed offspring mice to 12 mo of age after exposure to a maternal obesity and postnatal HFD. Both exposure to maternal obesity and postnatal HFD were independent risk factors for steatosis. At 12 mo, offspring exposed to both maternal obesity and postnatal HFD had histopathologic evidence of steatohepatitis and fibrosis, increased hepatic mRNA expression of genes coding for inflammatory cytokines such as interleukin-6 and tumor necrosis factor-α and fibrogenic enzymes such as collagen type 1-α2 and α-smooth muscle actin (55). There was a significant interaction between maternal and postweaning HFD diets in association with offspring steatohepatitis. Studies in NHP take this one step further. Evidence for early activation of Kupffer cells within the fetal liver is currently lacking; however, findings in the NHP suggest that maternal HFD can “prime” their proinflammatory response. Kupffer cells derived from juvenile offspring exposed to HFD during gestation, but currently on a healthy diet, have an exacerbated response when exposed to lipid or LPS prior to any observed development of obesity (Thorn SR, Newsom SA, Baquero KC et al., unpublished data). This priming may occur via upregulation of Toll-like receptor 4 in response to inflammatory stimuli, which has been demonstrated previously in macrophages (56,57).
Epigenetic Mechanisms of Intergenerational Transfer of Risk of NAFLD
There has been considerable progress in understanding epigenetic mechanisms by which the fetal liver might be “primed” by gestational overnutrition. Epigenetic modifications of DNA, such as methylation, acetylation, or histone modification, derive from gene–environment interactions that then influence transcription. Sookoian et al. demonstrated the importance of methylation of peroxisome proliferator–activated receptor γ coactivator 1α gene (PGC1α) in hepatic insulin resistance in NAFLD. PGC1α is involved in multiple aspects of energy metabolism in the liver, including insulin sensitivity, mitochondrial biogenesis, and oxidative phosphorylation. From liver tissue sampled from adult humans, there was increased methylation of the PGC1α promoter in NAFLD compared to controls. Methylation of PGC1α was positively correlated with peripheral insulin resistance and inversely correlated with hepatic PGC1α mRNA expression and hepatic mitochondrial content (58). The authors had previously shown a positive correlation between maternal prepregnancy BMI and methylation status of the PGC1α promoter in offspring umbilical cord blood (59). In an effort to link these two human findings, Pirola and colleagues returned to a murine model of maternal overnutrition. Maternal HFD led to a decrease in offspring hepatic mitochondrial content, as seen in adult humans with NAFLD, but the anticipated decrease in PGC1α mRNA expression and insulin resistance was only found in male offspring (60). The programming effect of maternal obesity on offspring mitochondrial function and metabolic control may not be organ specific and is evident in early embryogenesis and even in oocytes prior to fertilization (61,62).
Strakovsky et al. found increased expression of phosphoenolpyruvate kinase (PEPCK), the rate-limiting enzyme in the gluconeogenic pathway, in gestation 20-d-old fetal liver exposed to a maternal HFD. Further analysis demonstrated histone modifications of the phosphoenol pyruvate carboxy kinase 1 gene (PCK1) that codes for PEPCK, suggesting an epigenetic mechanism by which maternal diet could prime offspring liver for increased hepatic glucose production (63). In a separate mouse model of maternal HFD, Dudley et al. studied the impact of gestational overnutrition on offspring hepatocyte cell cycle dynamics, which potentially would impact hepatic resiliency in the face of injury or stress. The livers of offspring mice exposed to maternal HFD were hypomethylated at the Cdkn1a gene, which codes for a cell cycle inhibitory protein, and these livers appeared to have impaired regenerative capacity compared to controls (64). Maternal HFD also leads to histone modifications in the fetal livers of NHP (47). An additional mechanism by which maternal conditions might influence offspring gene expression is through alteration of offspring microRNA expression. A sheep model of maternal obesity showed reduced expression in fetal muscle of miRNA let-7g, which is involved in adipogenesis and inflammatory cytokine release (65). Hepatic expression of the micro RNA let-7c was reduced in the offspring of HFD-fed dams in a mouse model of maternal obesity (66). Although these data are suggestive, definitive evidence proving epigenetic inheritance as a risk for NAFLD is still lacking.
Microbiome and NAFLD
High-throughput sequencing technology has allowed for an explosion of research in the intestinal microbiome over the last decade, and there is great interest in understanding how the human intestine, sterile in utero, becomes colonized with bacterial loads that approach a density of 1010 colony-forming units per ml of colonic material. Studies in mother/infant pairs demonstrate that maternal vaginal and intestinal bacterial communities shape colonization patterns in the neonatal gut (67). The microbial composition of the infant intestine is highly variable and changes rapidly in early childhood, but as children get older, their intestinal microbiota begins to more closely resemble their mother’s (68,69,70,71). The characteristics of the inherited microbiome are likely very important in understanding offspring health as recent research suggests that aberrant metabolic phenotypes can be directly attributed to intestinal bacterial communities.
Functional differences in gut bacteria between obese and lean humans are apparent in experiments where fecal bacteria are transplanted from adult humans to germ-free mice. Germ-free mice receiving intestinal bacteria from obese humans will harvest calories more efficiently from food and experience an increase in % body fat compared to those mice receiving bacteria from lean humans (72). When newborn mice are exposed to antibiotics, there is disruption of intestinal colonization that results in increased energy harvest and adiposity in young mice (73). Metabolic substrates reaching the colon, including polysaccharides such as starch and fiber as well as protein, are fermented by intestinal bacteria leading to the release of short chain FA as a byproduct (74). At the level of the intestinal epithelium, short-chain FA appear to play a role in nutrient sensing and energy balance (75). Short-chain FA are absorbed into the portal circulation, reaching the liver where they are oxidized for cellular energy and/or feed metabolic pathways including ketone body production, lipogenesis, and gluconeogenesis. Certain bacteria are likely more adept at fermenting metabolic substrates that reach the colon, leading to more efficient energy harvest from food.
Le Roy et al. (76) recently used a mouse model to demonstrate a role for intestinal bacteria in creating a NAFLD-like phenotype. Intestinal bacteria, harvested from insulin-resistant mice on a HFD, were transplanted into germ-free animals. These germ-free animals were maintained on a HFD for 16 wk and then studied. When compared to control animals who had received their fecal transplants from insulin-sensitive mice, there were no differences in body weight and food consumption, yet those animals transplanted from insulin resistant mice had increased fasting glycemia and insulin levels as well as increased hepatic fat deposition. These mice had increased expression of transcription factors such as steroid regulatory element–binding protein and carbohydrate-responsive element binding protein that are key regulators of hepatic de novo lipogenesis( (76). If humans are endowed with intestinal bacteria that are more efficient in extracting calories, this would be a competitive advantage for an organism in an environment of food scarcity. However, in an environment of consistent food abundance, this metabolic efficiency may become deleterious to health. In mouse models, changes in the intestinal microbiota have been associated with hepatic inflammation and progression of steatohepatitis (77). As we further our understanding of the homeostatic relationships between ourselves and our commensal intestinal bacteria, it is exciting to consider that we may be able to manipulate our microbiota in ways that reduce the likelihood of NAFLD and other adverse metabolic outcomes.
Summary
Interest in how altered exposure to excess fuels affects early infant development has intensified in recent years due to recognition that pregnant women with obesity or gestational diabetes may transmit this phenotype to their offspring, leading to a vicious cycle of obesity and diabetes over generations. It now seems likely that NAFLD, which is the hepatic manifestation of the metabolic syndrome, shares a developmental origin that begins during gestation. Hypothesized mechanisms for intergenerational transmission of risk for NAFLD are summarized in Figure 2 .

Proposed pathways by which nonalcoholic fatty liver disease is programmed in offspring of mothers with prepregnancy obesity and insulin resistance. The gestational environment drives increased metabolic fuel delivery to the fetus, leading to fetal hepatic fat accumulation. Via epigenetic changes acquired prenatally, the liver is now “primed” for postnatal fat storage and inflammation mediated by Kupffer cells. A childhood high-fat diet then triggers further steatosis, hepatocyte injury, inflammation, and fibrosis via activation of hepatic stellate cells. DAG, diacylglycerol; TG, triglyceride.
Mounting evidence from laboratory and clinical studies focusing on the developmental basis of adult disease leads to the inescapable conclusion that greater focus is needed on the metabolic health of women prior to and during pregnancy. Data from the Japanese macaque model are encouraging. After being maintained on a HFD for 4 y, a group of female macaques was transitioned to a normal chow diet prior to breeding and during pregnancy. These diet-reversal mothers remained obese. However, offspring had substantial attenuation of steatosis and expression of genes involved in gluconeogenesis, suggesting that a dietary change during pregnancy can possibly mitigate the NAFLD phenotype in offspring of obese mothers (36). The contributions of excess maternal fuels, infant microbiome, inflammation, and the role of epigenetic changes in mitochondria are just beginning to be understood. However, with rapidly advancing tools such as transcriptomics, epigenetics, and metabolomics, along with expanding clinical investigation, it may be possible in the future to understand the earliest origins and biomarkers for NAFLD in mothers and infants. In the meantime, maintenance of normal maternal weight and insulin regulation at the outset of pregnancy is critical, as is attention to healthy diet and appropriate gestational weight gain, not just for ensuring good pregnancy outcomes but for the long-term health of offspring.
Statement of Financial Support
This study was supported by National Institutes of Health (Bethesda, MD): DK-090964, DK-077630, DK-078645, and DK-088324.
References
Agopian VG, Kaldas FM, Hong JC, et al. Liver transplantation for nonalcoholic steatohepatitis: the new epidemic. Ann Surg 2012;256:624–33.
Alisi A, Manco M, Vania A, Nobili V . Pediatric nonalcoholic fatty liver disease in 2009. J Pediatr 2009;155:469–74.
Welsh JA, Karpen S, Vos MB . Increasing prevalence of nonalcoholic Fatty liver disease among United States adolescents, 1988–1994 to 2007–2010. J Pediatr 2013;162:496–500 e1.
Feldstein AE, Charatcharoenwitthaya P, Treeprasertsuk S, Benson JT, Enders FB, Angulo P . The natural history of non-alcoholic fatty liver disease in children: a follow-up study for up to 20 years. Gut 2009;58:1538–44.
Schwimmer JB, McGreal N, Deutsch R, Finegold MJ, Lavine JE . Influence of gender, race, and ethnicity on suspected fatty liver in obese adolescents. Pediatrics 2005;115:e561–5.
Guerrero R, Vega GL, Grundy SM, Browning JD . Ethnic differences in hepatic steatosis: an insulin resistance paradox? Hepatology 2009;49:791–801.
Pacifico L, Cantisani V, Ricci P, et al. Nonalcoholic fatty liver disease and carotid atherosclerosis in children. Pediatr Res 2008;63:423–7.
Machado MV, Ferreira DM, Castro RE, et al. Liver and muscle in morbid obesity: the interplay of fatty liver and insulin resistance. PLoS ONE 2012;7:e31738.
Schwimmer JB, Pardee PE, Lavine JE, Blumkin AK, Cook S . Cardiovascular risk factors and the metabolic syndrome in pediatric nonalcoholic fatty liver disease. Circulation 2008;118:277–83.
Sundaram SS, Zeitler P, Nadeau K . The metabolic syndrome and nonalcoholic fatty liver disease in children. Curr Opin Pediatr 2009;21:529–35.
Schwimmer JB, Behling C, Newbury R, et al. Histopathology of pediatric nonalcoholic fatty liver disease. Hepatology 2005;42:641–9.
Brunt EM, Kleiner DE, Wilson LA, et al.; NASH Clinical Research NetworkA list of members of the Nonalcoholic Steatohepatitis Clinical Research Network can be found in the Appendix. Portal chronic inflammation in nonalcoholic fatty liver disease (NAFLD): a histologic marker of advanced NAFLD-Clinicopathologic correlations from the nonalcoholic steatohepatitis clinical research network. Hepatology 2009;49:809–20.
Kim SY, Dietz PM, England L, Morrow B, Callaghan WM . Trends in pre-pregnancy obesity in nine states, 1993-2003. Obesity (Silver Spring) 2007;15:986–93.
Dabelea D, Snell-Bergeon JK, Hartsfield CL, Bischoff KJ, Hamman RF, McDuffie RS ; Kaiser Permanente of Colorado GDM Screening Program. Increasing prevalence of gestational diabetes mellitus (GDM) over time and by birth cohort: Kaiser Permanente of Colorado GDM Screening Program. Diabetes Care 2005;28:579–84.
Hinkle SN, Sharma AJ, Kim SY, et al. Prepregnancy obesity trends among low-income women, United States, 1999-2008. Matern Child Health J 2012;16:1339–48.
Stamnes Køpp UM, Dahl-Jørgensen K, Stigum H, Frost Andersen L, Næss Ø, Nystad W . The associations between maternal pre-pregnancy body mass index or gestational weight change during pregnancy and body mass index of the child at 3 years of age. Int J Obes (Lond) 2012;36:1325–31.
Oken E, Taveras EM, Kleinman KP, Rich-Edwards JW, Gillman MW . Gestational weight gain and child adiposity at age 3 years. Am J Obstet Gynecol 2007;196:322.e1–8.
Whitaker RC . Predicting preschooler obesity at birth: the role of maternal obesity in early pregnancy. Pediatrics 2004;114:e29–36.
Dabelea D, Hanson RL, Lindsay RS, et al. Intrauterine exposure to diabetes conveys risks for type 2 diabetes and obesity: a study of discordant sibships. Diabetes 2000;49:2208–11.
Metzger BE, Lowe LP, Dyer AR, et al. Hyperglycemia and adverse pregnancy outcomes. N Engl J Med 2008;358:1991–2002.
Harmon KA, Gerard L, Jensen DR, et al. Continuous glucose profiles in obese and normal-weight pregnant women on a controlled diet: metabolic determinants of fetal growth. Diabetes Care 2011;34:2198–204.
Bildirici I, Roh CR, Schaiff WT, Lewkowski BM, Nelson DM, Sadovsky Y . The lipid droplet-associated protein adipophilin is expressed in human trophoblasts and is regulated by peroxisomal proliferator-activated receptor-gamma/retinoid X receptor. J Clin Endocrinol Metab 2003;88:6056–62.
Bernstein IM, Goran MI, Amini SB, Catalano PM . Differential growth of fetal tissues during the second half of pregnancy. Am J Obstet Gynecol 1997;176(1 Pt 1):28–32.
Herrera E, Amusquivar E . Lipid metabolism in the fetus and the newborn. Diabetes Metab Res Rev 2000;16:202–10.
Wells JC . Maternal capital and the metabolic ghetto: An evolutionary perspective on the transgenerational basis of health inequalities. Am J Hum Biol 2010;22:1–17.
Garg A . Clinical review#: Lipodystrophies: genetic and acquired body fat disorders. J Clin Endocrinol Metab 2011;96:3313–25.
Brumbaugh DE, Tearse P, Cree-Green M, et al. Intrahepatic fat is increased in the neonatal offspring of obese women with gestational diabetes. J Pediatr 2013;162:930–6.e1.
Modi N, Murgasova D, Ruager-Martin R, et al. The influence of maternal body mass index on infant adiposity and hepatic lipid content. Pediatr Res 2011;70:287–91.
Chen H, Morris MJ . Differential responses of orexigenic neuropeptides to fasting in offspring of obese mothers. Obesity (Silver Spring) 2009;17:1356–62.
Chang GQ, Gaysinskaya V, Karatayev O, Leibowitz SF . Maternal high-fat diet and fetal programming: increased proliferation of hypothalamic peptide-producing neurons that increase risk for overeating and obesity. J Neurosci 2008;28:12107–19.
Johnson W, Choh AC, Soloway LE, Czerwinski SA, Towne B, Demerath EW . Eighty-year trends in infant weight and length growth: the Fels Longitudinal Study. J Pediatr 2012;160:762–8.
Calegari VC, Torsoni AS, Vanzela EC, et al. Inflammation of the hypothalamus leads to defective pancreatic islet function. J Biol Chem 2011;286:12870–80.
Grant WF, Nicol LE, Thorn SR, Grove KL, Friedman JE, Marks DL . Perinatal exposure to a high-fat diet is associated with reduced hepatic sympathetic innervation in one-year old male Japanese macaques. PLoS ONE 2012;7:e48119.
Butte NF, Hopkinson JM, Wong WW, Smith EO, Ellis KJ . Body composition during the first 2 years of life: an updated reference. Pediatr Res 2000;47:578–85.
Frias AE, Morgan TK, Evans AE, et al. Maternal high-fat diet disturbs uteroplacental hemodynamics and increases the frequency of stillbirth in a nonhuman primate model of excess nutrition. Endocrinology 2011;152:2456–64.
McCurdy CE, Bishop JM, Williams SM, et al. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J Clin Invest 2009;119:323–35.
Dyer JS, Rosenfeld CR, Rice J, Rice M, Hardin DS . Insulin resistance in Hispanic large-for-gestational-age neonates at birth. J Clin Endocrinol Metab 2007;92:3836–43.
Catalano PM, Presley L, Minium J, Hauguel-de Mouzon S . Fetuses of obese mothers develop insulin resistance in utero. Diabetes Care 2009;32:1076–80.
Bruce KD, Cagampang FR, Argenton M, et al. Maternal high-fat feeding primes steatohepatitis in adult mice offspring, involving mitochondrial dysfunction and altered lipogenesis gene expression. Hepatology 2009;50:1796–808.
Bayol SA, Simbi BH, Fowkes RC, Stickland NC . A maternal “junk food” diet in pregnancy and lactation promotes nonalcoholic Fatty liver disease in rat offspring. Endocrinology 2010;151:1451–61.
Gregorio BM, Souza-Mello V, Carvalho JJ, Mandarim-de-Lacerda CA, Aguila MB . Maternal high-fat intake predisposes nonalcoholic fatty liver disease in C57BL/6 offspring. Am J Obstet Gynecol 2010;203:495.e1–8.
Shankar K, Kang P, Harrell A, et al. Maternal overweight programs insulin and adiponectin signaling in the offspring. Endocrinology 2010;151:2577–89.
Borengasser SJ, Lau F, Kang P, et al. Maternal obesity during gestation impairs fatty acid oxidation and mitochondrial SIRT3 expression in rat offspring at weaning. PLoS ONE 2011;6:e24068.
Kohjima M, Enjoji M, Higuchi N, et al. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int J Mol Med 2007;20:351–8.
Yeon JE, Choi KM, Baik SH, et al. Reduced expression of peroxisome proliferator-activated receptor-alpha may have an important role in the development of non-alcoholic fatty liver disease. J Gastroenterol Hepatol 2004;19:799–804.
Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, Li X . Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab 2009;9:327–38.
Suter MA, Chen A, Burdine MS, et al. A maternal high-fat diet modulates fetal SIRT1 histone and protein deacetylase activity in nonhuman primates. FASEB J 2012;26:5106–14.
Bouanane S, Merzouk H, Benkalfat NB, et al. Hepatic and very low-density lipoprotein fatty acids in obese offspring of overfed dams. Metab Clin Exp 2010;59:1701–9.
Alisi A, Feldstein AE, Villani A, Raponi M, Nobili V . Pediatric nonalcoholic fatty liver disease: a multidisciplinary approach. Nat Rev Gastroenterol Hepatol 2012;9:152–61.
Friedman SL . Liver fibrosis in 2012: Convergent pathways that cause hepatic fibrosis in NASH. Nat Rev Gastroenterol Hepatol 2013;10:71–2.
Malhi H, Gores GJ . Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Semin Liver Dis 2008;28:360–9.
Alkhouri N, Carter-Kent C, Feldstein AE . Apoptosis in nonalcoholic fatty liver disease: diagnostic and therapeutic implications. Expert Rev Gastroenterol Hepatol 2011;5:201–12.
Tamimi TI, Elgouhari HM, Alkhouri N, et al. An apoptosis panel for nonalcoholic steatohepatitis diagnosis. J Hepatol 2011;54:1224–9.
Vos MB, Barve S, Joshi-Barve S, Carew JD, Whitington PF, McClain CJ . Cytokeratin 18, a marker of cell death, is increased in children with suspected nonalcoholic fatty liver disease. J Pediatr Gastroenterol Nutr 2008;47:481–5.
Mouralidarane A, Soeda J, Visconti-Pugmire C, et al. Maternal obesity programs offspring nonalcoholic fatty liver disease by innate immune dysfunction in mice. Hepatology 2013;58:128–38.
De Nardo D, De Nardo CM, Nguyen T, Hamilton JA, Scholz GM . Signaling crosstalk during sequential TLR4 and TLR9 activation amplifies the inflammatory response of mouse macrophages. J Immunol 2009;183:8110–8.
Romics L Jr, Dolganiuc A, Kodys K, et al. Selective priming to Toll-like receptor 4 (TLR4), not TLR2, ligands by P. acnes involves up-regulation of MD-2 in mice. Hepatology 2004;40:555–64.
Sookoian S, Rosselli MS, Gemma C, et al. Epigenetic regulation of insulin resistance in nonalcoholic fatty liver disease: impact of liver methylation of the peroxisome proliferator-activated receptor ? coactivator 1a promoter. Hepatology 2010;52:1992–2000.
Gemma C, Sookoian S, Alvariñas J, et al. Maternal pregestational BMI is associated with methylation of the PPARGC1A promoter in newborns. Obesity (Silver Spring) 2009;17:1032–9.
Burgueño AL, Cabrerizo R, Gonzales Mansilla N, Sookoian S, Pirola CJ . Maternal high-fat intake during pregnancy programs metabolic-syndrome-related phenotypes through liver mitochondrial DNA copy number and transcriptional activity of liver PPARGC1A. J Nutr Biochem 2013;24:6–13.
Igosheva N, Abramov AY, Poston L, et al. Maternal diet-induced obesity alters mitochondrial activity and redox status in mouse oocytes and zygotes. PLoS ONE 2010;5:e10074.
Jungheim ES, Schoeller EL, Marquard KL, Louden ED, Schaffer JE, Moley KH . Diet-induced obesity model: abnormal oocytes and persistent growth abnormalities in the offspring. Endocrinology 2010;151:4039–46.
Strakovsky RS, Zhang X, Zhou D, Pan YX . Gestational high fat diet programs hepatic phosphoenolpyruvate carboxykinase gene expression and histone modification in neonatal offspring rats. J Physiol (Lond) 2011;589(Pt 11):2707–17.
Dudley KJ, Sloboda DM, Connor KL, Beltrand J, Vickers MH . Offspring of mothers fed a high fat diet display hepatic cell cycle inhibition and associated changes in gene expression and DNA methylation. PLoS ONE 2011;6:e21662.
Yan X, Huang Y, Zhao JX, et al. Maternal obesity downregulates microRNA let-7g expression, a possible mechanism for enhanced adipogenesis during ovine fetal skeletal muscle development. Int J Obes (Lond) 2013;37:568–75.
Zhang J, Zhang F, Didelot X, et al. Maternal high fat diet during pregnancy and lactation alters hepatic expression of insulin like growth factor-2 and key microRNAs in the adult offspring. BMC Genomics 2009;10:478.
Dominguez-Bello MG, Costello EK, Contreras M, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci USA 2010;107:11971–5.
Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO . Development of the human infant intestinal microbiota. PLoS Biol 2007;5:e177.
Koren O, Goodrich JK, Cullender TC, et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell 2012;150:470–80.
Turnbaugh PJ, Hamady M, Yatsunenko T, et al. A core gut microbiome in obese and lean twins. Nature 2009;457:480–4.
Yatsunenko T, Rey FE, Manary MJ, et al. Human gut microbiome viewed across age and geography. Nature 2012;486:222–7.
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI . An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006;444:1027–31.
Cho I, Yamanishi S, Cox L, et al. Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature 2012;488:621–6.
Schwiertz A, Taras D, Schäfer K, et al. Microbiota and SCFA in lean and overweight healthy subjects. Obesity (Silver Spring) 2010;18:190–5.
Tilg H, Kaser A . Gut microbiome, obesity, and metabolic dysfunction. J Clin Invest 2011;121:2126–32.
Le Roy T, Llopis M, Lepage P, et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut 2013;62:1787–94.
Henao-Mejia J, Elinav E, Jin C, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012;482:179–85.
Author information
Authors and Affiliations
Corresponding author
PowerPoint slides
Rights and permissions
About this article
Cite this article
Brumbaugh, D., Friedman, J. Developmental origins of nonalcoholic fatty liver disease. Pediatr Res 75, 140–147 (2014). https://doi.org/10.1038/pr.2013.193
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2013.193
This article is cited by
-
Prevalence of incidental sonographic findings of hepatic steatosis in children under 4 years of age
Pediatric Radiology (2023)
-
Murine neonatal ketogenesis preserves mitochondrial energetics by preventing protein hyperacetylation
Nature Metabolism (2021)
-
Maternal diet intervention before pregnancy primes offspring lipid metabolism in liver
Laboratory Investigation (2020)
-
Free-breathing 3-D quantification of infant body composition and hepatic fat using a stack-of-radial magnetic resonance imaging technique
Pediatric Radiology (2019)
-
DNA methylation patterns in peripheral blood mononuclear cells from Holstein cattle with variable milk yield
BMC Genomics (2018)
