
Abstract
The NLR family, pyrin domain-containing 3 (NLRP3) inflammasome is a multiprotein complex that activates caspase 1, leading to the processing and secretion of the pro-inflammatory cytokines interleukin-1β (IL-1β) and IL-18. The NLRP3 inflammasome is activated by a wide range of danger signals that derive not only from microorganisms but also from metabolic dysregulation. It is unclear how these highly varied stress signals can be detected by a single inflammasome. In this Opinion article, we review the different signalling pathways that have been proposed to engage the NLRP3 inflammasome and suggest a model in which one of the crucial elements for NLRP3 activation is the generation of reactive oxygen species (ROS).
Similar content being viewed by others

Main
Multicellular organisms, from plants to vertebrates, are constantly subject to environmental, intracellular and extracellular insults. Examples of such insults include pathogens, DNA damage and metabolic stress. To mount an appropriate repair response to these insults, the organism requires mechanisms that identify the source of the problem and engage suitable effectors. Cellular insult is identified by the innate immune system through various cellular receptors that orchestrate protection and repair mechanisms — such as the nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), the retinoic acid-inducible gene I-like receptors (RLRs) and the Toll-like receptors (TLRs). Often, these pathways are successful; however, if the source of cellular stress cannot be resolved, damaged cells are committed to suicide by apoptosis or necrosis and targeted for removal by specialized immune cells. Here, we review the proposed activation mechanisms of one such receptor responsible for recognizing cellular stress. NLR family, pyrin domain-containing 3 (NLRP3; also known as NALP3 or cryopyrin) was recently identified to form a cytoplasmic complex known as the NLRP3 inflammasome, which potently modulates innate immune function by regulating the maturation and secretion of pro-inflammatory cytokines, such as interleukin-1β (IL-1β). The finding that hyperactivity of this complex underlies several human diseases, including Muckle–Wells syndrome (a type of cryopyrin-associated periodic syndrome), emphasizes the importance of this pathway. A precise understanding of the signals that activate the NLRP3 inflammasome is therefore crucial for the development of new anti-inflammatory drugs.
Stress signalling is evolutionarily conserved
Given that the range of potential cellular insults has not changed markedly during evolution, it is not surprising that the pathways triggered by cellular stress situations are also highly conserved. Plants respond to pathogens or metabolic stress by rapidly activating a battery of defence responses1,2,3,4. For example, pathogen invasion of a resistant plant can result in macroscopic lesions that form as a result of cellular necrosis termed the 'hypersensitive response'. The hypersensitive response is associated with several physiological changes, including transient opening of ion channels, in particular Ca2+ and K+ channels, and/or the production of reactive oxygen species (ROS). Similarly in mammals, pathogen recognition by receptors of the innate immune system, such as TLRs and NLRs, triggers a range of inflammatory responses, often including the generation of ROS and the modulation of Ca2+ and K+ ion fluxes (reviewed in Ref. 5).
Stress-induced ROS function as an alarm signal that triggers efficient defence responses by modulating specific signal transduction pathways that use hydrogen peroxide as a secondary messenger, such as the pathway leading to the NFE2-related factor 2 (NRF2)-mediated antioxidant response6. ROS activity is rigorously controlled by a versatile antioxidant system that modulates intracellular ROS concentration. Nonetheless, if stress is prolonged, ROS concentrations can overcome the scavenging action of the antioxidant system, resulting in extensive cellular damage and necrosis.
In plants, resistance (R) genes are implicated in ROS sensing and generation and are crucial for plant defences, including the hypersensitivity response, against pathogens and other stressors1,2,3,4. The largest class of R genes encodes cytoplasmic proteins with structural homology to mammalian stress sensors, the NLRs. Both NLRs and R proteins have carboxy-terminal leucine-rich repeats (LRRs), a central oligomerization module (NB-ARC or domain present in NAIP, CIITA, HET-E and TP1 (NACHT)) and an amino-terminal effector domain5. The N terminus of plant R proteins often contains a Toll/interleukin-1 receptor (TIR) domain; in mammals, the TIR domain is a well-known protein interaction motif that transduces signalling induced by IL-1 receptor (IL-1R) and TLRs5. In humans, common NLR N-terminal effector domains include pyrin domain (PYD), caspase-recruitment domain (CARD) and baculovirus inhibitor of apoptosis protein repeat (BIR)7, which have presumably evolved to enable connections to new signalling pathways. For example, NLRP3 has an N-terminal PYD that enables the assembly of the inflammasome complex.
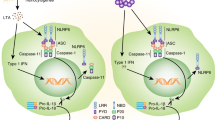
Consistent with the structural similarity of stress-sensing receptors between plants and mammals, the signalling pathways engaged by these receptors are also similar. In both plants and mammals, stress signalling follows the same general scheme: first, stress situations are detected directly or indirectly by a sensor protein (such as an NLR or R protein); second, subsequent oligomerization of the sensor allows the recruitment of effector proteins; and finally, activation of effector proteins engages cellular repair responses. In mammals, the archetypal example of this activation scheme is the formation of the NLRP3 inflammasome during early innate immune responses to pathogens or endogenous danger signals. The NLRP3 inflammasome contains NLRP3, the adaptor protein apoptosis-associated speck-like protein containing a CARD (ASC; also known as PYCARD) and the effector cysteine protease caspase 1 (Ref. 8). Following detection of cellular stress, NLRP3 oligomerizes through homotypic interactions between NACHT domains (Fig. 1). The PYD of NLRP3 is then exposed for interaction with the PYD of ASC. The CARD of ASC in turn recruits pro-caspase 1 through CARD–CARD interactions. Pro-caspase 1 clustering on oligomerized NLRP3 results in caspase 1 auto-activation and caspase 1-dependent processing of cytoplasmic targets, including the pro-inflammatory cytokines IL-1β and IL-18, which mediate repair responses such as angiogenesis and neutrophil influx to remove cellular debris. The mature cytokines are released from the cell by an unconventional secretion pathway that is currently not defined9. ASC and caspase 1 are co-secreted with IL-1β and IL-18 (Ref. 10), but the extracellular functions of ASC and caspase 1, if any, are not known.

Under healthy cellular conditions, NLR family, pyrin domain-containing 3 (NLRP3) is auto-repressed owing to an internal interaction between the NACHT domain and LRRs. This auto-repression is removed in the presence of pathogen-associated molecular patterns (PAMPs) from microorganisms or damage-associated molecular patterns (DAMPs) from endogenous danger signals. This results in exposure of the NACHT domain. In turn, NLRP3 oligomerizes and recruits apoptosis-associated speck-like protein containing a CARD (ASC; also known as PYCARD) and pro-caspase 1, triggering the activation of caspase 1 and the maturation and secretion of pro-inflammatory cytokines such as interleukin-1β (IL-1β) and IL-18. Other cytoplasmic proteins, such as enzymes of the glycolytic pathway, are also substrates of active caspase 1. CARD, caspase-recruitment domain; LRRs, leucine-rich repeats; NACHT, NAIP, CIITA, HET-E and TP1; PYD, pyrin domain.
Activators of the NLRP3 inflammasome
Various danger signals activate the NLRP3 inflammasome (Table 1). These include whole pathogens, pathogen-associated molecular patterns (PAMPs) and other pathogen-associated molecules (such as bacterial pore-forming toxins and the malaria parasite product haemozoin). Environmental irritants (such as asbestos) and damage-associated molecular patterns (DAMPs), which are host-derived molecules that are indicative of cellular damage (such as extracellular ATP), also activate the NLRP3 inflammasome. The mechanisms by which these structurally distinct molecules trigger NLRP3 oligomerization and inflammasome activation are currently unclear and have been intensely debated in recent literature7. However, all of the proposed models agree that cytoplasmic K+ concentration crucially affects inflammasome activation, and K+ efflux from the cell should be factored into any proposed scheme for NLRP3 inflammasome activation. Cytoplasmic K+ concentration in healthy cells is ∼140–150 mM, a concentration that does not allow NLRP3 activation. ATP, a potent activator of the NLRP3 inflammasome, decreases intracellular K+ concentration by approximately 50%11. Accordingly, a shift in cytoplasmic K+ concentration to less than 70 mM is required for NLRP3 inflammasome activity in vitro12. Inhibition of K+ efflux by high extracellular K+ concentration blocks inflammasome activation in response to the imiquimod R837, the gout-associated crystal monosodium urate (MSU), the bacterial ionophore nigericin, ATP, asbestos, malarial haemozoin and Candida albicans (reviewed in Ref. 5). Interestingly, low cytoplasmic K+ concentration is also required for the activation of the apoptosome (the caspase-containing protein complex that forms during the process of apoptosis), which indicates that 'normal' intracellular concentrations of K+ safeguard the cell against inappropriate formation of caspase-containing stress complexes in general13. Although the mechanism by which cytoplasmic K+ concentration modulates NLRP3 inflammasome activation is unknown, it seems that intracellular K+ depletion alone is not sufficient for NLRP3-mediated IL-1β processing and secretion14.
NLRP3 inflammasome activation models
Three models for activation of the NLRP3 inflammasome have been proposed.
The channel model. Extracellular ATP is a NLRP3 agonist that is released at sites of cellular injury or necrosis. ATP-mediated inflammasome activation depends on activation of the P2X7 ATP-gated ion channel, which triggers rapid K+ efflux from the cell15 and gradual recruitment and pore formation of the pannexin 1 hemichannel14,16. Pannexin 1 is a gap junction protein, but it does not seem to form functional gap junctions in vivo. Instead, pannexin 1 functions as a membrane channel that carries ions and signalling molecules between the cytoplasm and the extracellular space. As such, it is a candidate ATP release channel in various cell types17. One model of inflammasome activation proposes that P2X7-dependent formation of the pannexin 1 hemichannel allows extracellular NLRP3 activators, in particular bacterial products, to access the cytoplasm and to interact with and activate NLRP3 directly16 (Fig. 2). In support of such a model, bacterial muramyl dipeptide (MDP) that has been phagocytosed translocates from acidified phagocytic vesicles into the cytoplasm through a pathway that depends on pannexin 1 function18. Moreover, heat killed bacteria activate the NLRP3 inflammasome when artificially delivered to the cytoplasm by the streptococcal pore-forming protein streptolysin O19. Pore formation as described in this model also provides an explanation for the potent NLRP3-activating properties of bacterial pore-forming proteins such as the α-toxin of Staphyloccocus aureus10. Any pore in the plasma membrane would also automatically lead to K+ efflux from the cell, which is a requirement for inflammasome activation, adding to the appeal of this model. However, so far there have been no reports showing direct interaction between NLRP3 and an inflammasome agonist, and, given the structural diversity of activating stimuli, it is difficult to imagine that NLRP3 might sense more than a few of its activating extracellular stimuli directly. Moreover, some NLRP3 agonists, such as MSU crystals or the particulate asbestos, are too large for cytoplasmic translocation through any type of channel or pore. So, although a specific subset of NLRP3 agonists could enter cells through membrane pores and bind directly to NLRP3, this model cannot account for NLRP3 inflammasome activity in response to all stimuli.

The NLR family, pyrin domain-containing 3 (NLRP3) inflammasome is activated by extracellular ATP, which stimulates the P2X7 K+ release channel, which in turn leads to formation of the associated pannexin 1 pore. This pore, as well as pores formed by bacterial toxins, allows cytoplasmic entry of extracellular factors that are direct NLRP3 ligands and also allows K+ efflux from the cell. ASC, apoptosis-associated speck-like protein containing a CARD; DAMPs, damage-associated molecular patterns; IL, interleukin; PAMPs, pathogen-associated molecular patterns.
The lysosome rupture model. An alternative model of inflammasome activation that takes into account the size of the activators is the lysosome rupture model20,21. According to this model, which is particularly useful for explaining NLRP3 inflammasome activation by large particulate activators (such as alum and silica), inefficient clearance of the activating particle following phagocytosis leads to phagosomal destabilization and lysosome rupture (Fig. 3). The ensuing release of the lysosomal protein cathepsin B into the cytoplasm triggers inflammasome activation directly or indirectly through an uncharacterized pathway. This model is supported by observations that cathepsin B inactivation in human cells by a cathepsin B inhibitor impairs NLRP3 inflammasome activation in response to particulate activators and that artificial lysosome disruption is sufficient for spontaneous NLRP3 activity20. However, it is still unclear whether this signalling pathway is functional in mice. Initial results showing decreased IL-1β secretion by macrophages derived from mice deficient in cathepsin B21 could not be confirmed by others22. This might indicate that this inflammasome activation pathway is restricted to humans or involves other cathepsins in mice; it is also possible that the cathepsin B inhibitor suppresses NLRP3 activation in humans through an off-target effect on a component of the NLRP3 inflammasome cascade other than cathepsin B, as recently suggested for the effects of a cathepsin B inhibitor on NALP1 inflammasome activity23.

Crystalline or particulate structures (such as alum and silica) are phagocytosed, leading to lysosome rupture and the cytoplasmic release of cathepsin B. Cathepsin B, either directly or through cleaving an unidentified substrate, induces activation of the NLR family, pyrin domain-containing 3 (NLRP3) inflammasome. ASC, apoptosis-associated speck-like protein containing a CARD; DAMPs, damage-associated molecular patterns; IL, interleukin; PAMPs, pathogen-associated molecular patterns.
The ROS model. A third model proposes that NLRP3 is a more general sensor of cellular stress through its activation by ROS generated in spatial and temporal proximity to the inflammasome12,24. All NLRP3 activators that have been examined, including ATP and particulate activators such as asbestos and silica (thus including activators that trigger channel formation and that cause lysosome rupture), trigger the generation of short-lived ROS, and treatment with various ROS scavengers blocks NLRP3 activation in response to a range of agonists12,25. ROS generation is frequently accompanied by K+ efflux26; the interplay between these pathways is currently unclear, but it is possible that low intracellular K+ concentration triggers ROS production or vice versa. ROS-mediated inflammasome activation and K+ efflux for agonists other than ATP are independent of the channel activity of P2X7 (Ref. 14).
A recent report gives some insight into the molecular events potentially driving ROS-dependent inflammasome activation24. Treatment with NLRP3 agonists triggers the association of NLRP3 with thioredoxin-interacting protein (TXNIP; also known as VDUP1), in a ROS-dependent manner. In unstimulated cells, TXNIP is constitutively bound to and inhibited by the oxidoreductase thioredoxin. Following an increase in cellular ROS concentration, this complex dissociates and TXNIP binds to NLRP3 (mainly to the LRRs), leading to NLRP3 activation (Fig. 4). In support of such an activation mechanism, knockdown of thioredoxin potentiates inflammasome activation24,25. Furthermore, TXNIP knockout or knockdown impairs caspase 1 activation and IL-1β secretion in macrophages following stimulation by various NLRP3 agonists, including R837, particulates such as MSU, alum or silica, and ATP24. Notably, capase 1 activation is not blocked completely in the absence of TXNIP, which indicates that other regulators of inflammasome activity exist and that other pathways might function together with the ROS pathway to initiate a complete inflammatory response.

All NLR family, pyrin domain-containing 3 (NLRP3) agonists that have been tested trigger the production of reactive oxygen species (ROS). ROS production results in NLRP3 inflammasome activation through release of the ROS-sensitive NLRP3 ligand thioredoxin-interacting protein (TXNIP) from its inhibitor thioredoxin (TRX). ASC, apoptosis-associated speck-like protein containing a CARD; DAMPs, damage-associated molecular patterns; IL, interleukin; PAMPs, pathogen-associated molecular patterns.
An integrated model of NLRP3 action?
Although our current understanding of the signalling pathways that direct NLRP3 inflammasome activation is rudimentary, we think that ROS are crucially involved, which is consistent with their evolutionarily ancient role in plant defence responses. However, many aspects of ROS-dependent inflammasome activation remain unknown (Box 1). For example, the source of ROS that activate the inflammasome is still uncharacterized. NADPH oxidases are a potential source of ROS in professional phagocytes, and they are most likely to function in ROS-dependent inflammasome activation by particulate agonists. In this case, 'frustrated phagocytosis' caused by the inefficient clearance of phagocytosed material is likely to ultimately result in chronic activation of NADPH oxidases and excessive ROS production25. At least seven NADPH oxidase complexes exist, five of which have a common P22PHOX (also known as CYBA) subunit27. Knockdown of P22PHOX impaired NLRP3 inflammasome activation by many stimuli25. However, macrophages deficient in subunits specific to three individual NADPH oxidase complexes (NOX1, NOX2 and NOX4) respond normally to inflammasome activators (C. Dostert and J.T., unpublished observations), which indicates that another P22PHOX-dependent oxidase might be responsible for generating NLRP3-activating ROS or that there is functional redundancy between NAPDH oxidase complexes. Alternatively, other sources of cellular ROS, such as mitochondria or xanthine oxidase, cannot be excluded.
Is it possible to link the ROS model to the other two proposed signalling pathways? The activation of NLRP3 by particulates might be explained by both lysosome rupture and ROS models; phagocytosed particulates that are too large to be efficiently cleared are likely to induce the production of ROS on their way to lysosomes, where they cause the rupture of the organelle. So, the lysosome rupture model could be viewed as forming part of a more general ROS pathway. Phagocytosis, leading to lysosome rupture, would be essential for ROS generation by particulates, whereas smaller activators such as ATP that do not need to be phagocytosed could induce the production of ROS differently, for example through the activation of the P2X7 receptor.
However, it is more difficult to integrate the channel model with the ROS or lysosome rupture models, as they constitute different fundamental mechanisms. Whereas the channel model proposes the physical entry of activators into the cytoplasm to allow direct contact with NLRP3, neither the ROS nor lysosome rupture models propose such a mechanism; instead, they posit that the NLRP3 inflammasome is activated through signals that indicate intracellular stress, such as ROS or cytoplasmic cathepsin B, respectively. This does not mean that the channel model is not important for inflammasome activation in response to certain stimuli.
Future directions
Future studies are required to address the many unresolved questions (Box 1) in this field, as further insight into the signalling pathways leading to NLRP3 inflammasome activation will be important for the development of new therapeutic anti-inflammatory drugs. The potential for therapeutics targeting this pathway is exemplified by the recent success of IL-1 antagonists for the treatment of patients with cryopyrin-associated periodic syndromes or gout (in which deposition of MSU crystals in the joints is thought to trigger inflammasome activation)28.
References
Hammond-Kosack, K. E. & Jones, J. D. Resistance gene-dependent plant defense responses. Plant Cell 8, 1773–1791 (1996).
Cohn, J., Sessa, G. & Martin, G. B. Innate immunity in plants. Curr. Opin. Immunol. 13, 55–62 (2001).
Lam, E. Controlled cell death, plant survival and development. Nature Rev. Mol. Cell Biol. 5, 305–315 (2004).
Miller, G. et al. The plant NADPH oxidase RBOHD mediates rapid systemic signaling in response to diverse stimuli. Sci. Signal. 2, ra45 (2009).
Martinon, F., Mayor, A. & Tschopp, J. The inflammasomes: guardians of the body. Annu. Rev. Immunol. 27, 229–265 (2009).
Adibhatla, R. M. & Hatcher, J. F. Lipid oxidation and peroxidation in CNS health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 12, 125–169 (2010).
Bryant, C. & Fitzgerald, K. A. Molecular mechanisms involved in inflammasome activation. Trends Cell Biol. 19, 455–464 (2009).
Agostini, L. et al. NALP3 forms an IL-1β processing inflammasome with increased activity in Muckle–Wells auto-inflammatory disorder. Immunity 20, 319–325 (2004).
Eder, C. Mechanisms of interleukin-1β release. Immunobiology 214, 543–553 (2009).
Mariathasan, S. et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440, 228–232 (2006).
Perregaux, D. & Gabel, C. A. Interleukin-1β maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J. Biol. Chem. 269, 15195–15203 (1994).
Pétrilli, V. et al. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 14, 1583–1589 (2007).
Cain, K., Langlais, C., Sun, X. M., Brown, D. G. & Cohen, G. M. Physiological concentrations of K+ inhibit cytochrome c-dependent formation of the apoptosome. J. Biol. Chem. 276, 41985–41990 (2001).
Pelegrin, P. & Surprenant, A. Pannexin-1 mediates large pore formation and interleukin-1β release by the ATP-gated P2X7 receptor. EMBO J. 25, 5071–5082 (2006).
Ferrari, D. et al. The P2X7 receptor: a key player in IL-1 processing and release. J. Immunol. 176, 3877–3883 (2006).
Kanneganti, T.-D., Lamkanfi, M. & Nunez, G. Intracellular NOD-like receptors in host defense and disease. Immunity 27, 549–559 (2007).
Sohl, G., Maxeiner, S. & Willecke, K. Expression and functions of neuronal gap junctions. Nature Rev. Neurosci. 6, 191–200 (2005).
Marina-García, N. et al. Pannexin-1-mediated intracellular delivery of muramyl dipeptide induces caspase-1 activation via cryopyrin/NLRP3 independently of Nod2. J. Immunol. 180, 4050–4057 (2008).
Kanneganti, T.-D. et al. Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity 26, 433–443 (2007).
Hornung, V. et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nature Immunol. 9, 847–856 (2008).
Halle, A. et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nature Immunol. 9, 857–865 (2008).
Dostert, C. et al. Malarial hemozoin is a Nalp3 inflammasome activating danger signal. PLoS ONE 4, e6510 (2009).
Newman, Z. L., Leppla, S. H. & Moayeri, M. CA-074Me protection against anthrax lethal toxin. Infect. Immun. 77, 4327–4336 (2009).
Zhou, R., Tardivel, A., Thorens, B., Choi, I. & Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nature Immunol. 11, 136–140 (2010).
Dostert, C. et al. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320, 674–677 (2008).
Kowaltowski, A. J., de Souza-Pinto, N. C., Castilho, R. F. & Vercesi, A. E. Mitochondria and reactive oxygen species. Free Radic. Biol. Med. 47, 333–343 (2009).
Krause, K.-H. & Bedard, K. NOX enzymes in immuno-inflammatory pathologies. Semin. Immunopathol. 30, 193–194 (2008).
McDermott, M. F. & Tschopp, J. From inflammasomes to fevers, crystals and hypertension: how basic research explains inflammatory diseases. Trends Mol. Med. 13, 381–388 (2007).
Walev, I., Reske, K., Palmer, M., Valeva, A. & Bhakdi, S. Potassium-inhibited processing of IL-1β in human monocytes. EMBO J. 14, 1607–1614 (1995).
Meissner, F., Molawi, K. & Zychlinsky, A. Superoxide dismutase 1 regulates caspase-1 and endotoxic shock. Nature Immunol. 9, 866–872 (2008).
Kanneganti, T.-D. et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 440, 233–236 (2006).
Allen, I. C. et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 30, 556–565 (2009).
Joly, S. et al. Cutting edge: Candida albicans hyphae formation triggers activation of the Nlrp3 inflammasome. J. Immunol. 183, 3578–3581 (2009).
Warren, S. E., Mao, D. P., Rodriguez, A. E., Miao, E. A. & Aderem, A. Multiple Nod-like receptors activate caspase 1 during Listeria monocytogenes infection. J. Immunol. 180, 7558–7564 (2008).
Yamasaki, K. et al. NLRP3/cryopyrin is necessary for interleukin-1β (IL-1β) release in response to hyaluronan, an endogenous trigger of inflammation in response to injury. J. Biol. Chem. 284, 12762–12771 (2009).
Cassel, S. L. et al. The Nalp3 inflammasome is essential for the development of silicosis. Proc. Natl Acad. Sci. USA 105, 9035–9040 (2008).
Kool, M. et al. Cutting edge: alum adjuvant stimulates inflammatory dendritic cells through activation of the NALP3 inflammasome. J. Immunol. 181, 3755–3759 (2008).
Acknowledgements
J.T. is supported by grants from the Swiss National Science Foundation, by EU grants Mugen, Hermione, Apo-Sys and Apo-Train and by the Institute of Arthritis Research. K.S. is supported by a CJ Martin Fellowship from the Australian National Health and Medical Research Council (ID 490993).
Author information
Authors and Affiliations
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Related links
Rights and permissions
About this article
Cite this article
Tschopp, J., Schroder, K. NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production?. Nat Rev Immunol 10, 210–215 (2010). https://doi.org/10.1038/nri2725
Published:
Issue Date:
DOI: https://doi.org/10.1038/nri2725
This article is cited by
-
Dual-Atom Nanozyme Eye Drops Attenuate Inflammation and Break the Vicious Cycle in Dry Eye Disease
Nano-Micro Letters (2024)
-
Effects of evodiamine on ROS/TXNIP/NLRP3 pathway against gouty arthritis
Naunyn-Schmiedeberg's Archives of Pharmacology (2024)
-
Adiponectin inhibits ROS/NLRP3 inflammatory pathway through FOXO3A to ameliorate oral submucosal fibrosis
Odontology (2024)
-
Verapamil-loaded supramolecular hydrogel patch attenuates metabolic dysfunction-associated fatty liver disease via restoration of autophagic clearance of aggregated proteins and inhibition of NLRP3
Biomaterials Research (2023)
-
Hypoxia exacerbates intestinal injury and inflammatory response mediated by myeloperoxidase during Salmonella Typhimurium infection in mice
Gut Pathogens (2023)
