
Key Points
-
The list of known variants affecting type 2 diabetes mellitus (T2DM) risk confirms that this disease has a multifactorial aetiology
-
The concept of precision medicine has been exemplified in pharmacogenetic studies of monogenic diabetes mellitus
-
The genetic architecture of mild adverse drug reactions and treatment efficacy for antidiabetic agents probably resembles that of T2DM and other complex traits
-
Existing pharmacogenetic evidence of T2DM is limited; future pharmacogenomic studies utilizing large samples sizes will help identify variants that reveal novel mechanisms of drug action
-
Genetic evidence-based 'dose-response' curves have been used in validating candidate drug targets
-
Pharmacogenomic studies adopting a systems biology approach are expected to provide context specific evidence for future T2DM drug development
Abstract
Genomic studies have greatly advanced our understanding of the multifactorial aetiology of type 2 diabetes mellitus (T2DM) as well as the multiple subtypes of monogenic diabetes mellitus. In this Review, we discuss the existing pharmacogenetic evidence in both monogenic diabetes mellitus and T2DM. We highlight mechanistic insights from the study of adverse effects and the efficacy of antidiabetic drugs. The identification of extreme sulfonylurea sensitivity in patients with diabetes mellitus owing to heterozygous mutations in HNF1A represents a clear example of how pharmacogenetics can direct patient care. However, pharmacogenomic studies of response to antidiabetic drugs in T2DM has yet to be translated into clinical practice, although some moderate genetic effects have now been described that merit follow-up in trials in which patients are selected according to genotype. We also discuss how future pharmacogenomic findings could provide insights into treatment response in diabetes mellitus that, in addition to other areas of human genetics, facilitates drug discovery and drug development for T2DM.
Similar content being viewed by others

Main
In the past decade, genome-wide association studies (GWAS) and high-throughput sequencing, propelled by the fast development in affordable genomic technologies, have greatly advanced our understanding of the genetic aetiology of many common diseases1. Pharmacogenomic studies applying these genome-wide approaches to investigate drug responses have also yielded important results2,3. In this Review, and in this context, we discuss the genomic evidence that has strengthened our understanding of the multifactorial aetiology of type 2 diabetes mellitus (T2DM) and discuss the emerging evidence that a complex genetic architecture might underline the variation in response to antidiabetic drugs. Genetic evidence in disease genomics is increasingly being used for target validation in drug discovery. We anticipate how robust pharmacogenomic evidence could provide valuable information for predicting both on-target and off-target effects in drug discovery and development.
The multifactorial aetiology of T2DM
T2DM is a complex metabolic disease characterized by hyperglycaemia resulting from functional impairment in insulin secretion, insulin action or both4. Both insulin resistance and secretory deficiency arise through the interplay of genetic and environmental risk factors5. GWAS, which have interrogated all the common genetic variants (minor allele frequency >5%), have identified >120 T2DM risk loci6,7. High-throughput sequencing studies, which could theoretically examine all the variants in the genome, or at least the section that encodes proteins, have also enabled the discovery of rare variants (minor allele frequency <5%) at GWAS-identified loci and novel loci for T2DM8,9. Together these common variants with small to moderate effects and rare variants with relatively large effects could account for ∼15% of the total risk of developing T2DM and confirm its nature as a multisystem disorder6,10.
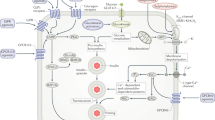
Glycaemic control is a key focus in the management of T2DM, and is associated with both microvascular and macrovascular benefits11,12,13. The treatment of T2DM has evolved with our understanding of the pathophysiology of this complex disease5. A wide range of drug treatments, characterized by different mechanisms of action, are available to achieve glycaemic control in patients with T2DM14 (Fig. 1). Apart from insulin replacement, traditional oral agents include secretagogues that stimulate the pancreas to release insulin and sensitizers that enhance the efficacy of insulin action14. New agents include dipeptidylpeptidase 4 (DPP-4) inhibitors, also known as the gliptins, that enhance the so-called 'incretin effect' and promote glucose-stimulated insulin secretion15; as well as sodium–glucose cotransporter 2 (SGLT-2) inhibitors that reduce hyperglycaemia by increasing glucose elimination via the urine16. Although these drugs are all effective at lowering glucose in patients with T2DM, glycaemic control often fails even after a combination of the available treatment options due to the progressive nature of the disease.

The mechanism for metformin action remains uncertain: metformin might target the liver to reduce gluconeogenesis and skeletal muscles to enhance peripheral glucose utilization110, with a possible role in the gut to increase levels of glucagon-like peptide 1 (GLP-1) (Ref. 111). Sulfonylureas and meglitinides increase insulin secretion in the pancreas112,113. Thiazolidinediones (TZDs) act as insulin sensitizers in skeletal muscle, adipose tissue and the liver114. GLP-1 receptor (GLP-1R) agonists (GLP-1RA) target the pancreas to increase insulin secretion and reduce glucagon production, as well as act in the gut to reduce gastric emptying115. Dipeptidyl peptidase 4 (DPP-4) inhibitors (DPP-4i) increase endogenous incretin levels by blocking the action of DPP-4 (Ref. 115). Sodium–glucose cotransporter 2 (SGLT-2) inhibitors (SGLT-2i) reduce renal glucose reabsorption116.
Antidiabetic drug responses can be considered at many levels, as outlined in Table 1, including the physiological response to the drug, or the long-term effect of the drug in terms of microvascular or macrovascular risk reduction. In this Review, when considering drug response, we focus primarily on the glycaemic effect of drugs as this outcome has been the most studied.
Monogenic diabetes mellitus
With the increasing awareness that T2DM is highly heterogeneous, and as we understand more about the aetiology of the disease, we can begin to subdivide T2DM into distinct aetiological subtypes. This development can be seen with the increasing identification of monogenic forms of the disease, which until the past 10–15 years were misclassified as type 1 diabetes mellitus or T2DM. Understanding these aetiological subtypes has resulted in some of the most clinically robust examples of pharmacogenetics to date. For example, patients with Maturity Onset Diabetes of the Young owing to mutations in HNF1A (which accounts for ∼3% of all diabetes mellitus cases diagnosed under the age of 30 years) are extremely sensitive to sulfonylurea treatment, and can successfully transition off insulin treatment17. Similarly, patients with neonatal diabetes mellitus due to KCNJ11 or ABCC8 mutations who have insulin-dependent diabetes mellitus from soon after birth have been shown to respond to high dose sulfonylureas and to be able to transition off insulin onto oral sulfonylurea treatment18. These examples highlight how increasing awareness of aetiological subtypes of diabetes mellitus will enable a precise approach to treatment of the disease and is an area of great interest. However, for the remainder of this Review, we focus on polygenic influences on drug responses in T2DM.
Pharmacogenomics and genetic architecture
Pharmacogenetics aims to seek the genetic explanation of why individuals respond differently to drugs, both in terms of therapeutic efficacy as well as adverse drug reactions (ADR)19. Before the emergence of genome-wide genotyping arrays, pharmacogenetic studies focused on candidate genes with known links to drug distribution, metabolism or response pathways19. With the development of cost-effective genomic technologies, genome-wide genotyping and sequencing has transformed this traditional pharmacogenetic approach into a more global pharmacogenomic approach that can systematically interrogate millions of genetic polymorphisms across the genome20,21. Most published genome-wide studies of drug response are GWAS, with only a few studies reporting sequencing-based investigations. One example of a sequencing-based study is the use of publicly available whole-genome sequence data on 482 samples to profile 231 genes22. In that study, the investigators also performed whole-genome sequencing on seven family members to try to explain the genetic basis of their variable response to anticoagulation treatment. The terms pharmacogenetics and pharmacogenomics are often used interchangeably, but in this Review we use pharmacogenomics to refer to studies using genome-wide approaches.
Biomarker discovery for precision medicine remains the long-term goal of pharmacogenomic studies. However, an often under-appreciated benefit of such studies is that they can advance our understanding of the biological mechanisms of drug action in humans by identifying variants in genes not previously thought to be associated with drug response. These genes might never have been included in traditional candidate gene approaches3.
A fundamental issue underlying the validity and feasibility of pharmacogenomic studies is the genetic architecture of drug response23. In this context 'genetic architecture' refers to the number of response variants; the frequency spectrum of these response variants; the effect-size spectrum of the variants; the physical distribution of the variants in the genome; and the amount of variation in drug response explained by these genetic variants (known as heritability). While heritability determines the validity of pharmacogenomic studies, the other aspects of genetic architecture dictate the feasibility and design of pharmacogenomic studies.
Adopting traditional twin and family study designs to estimate the heritability of drug treatment outcomes has been largely impractical, because family members might not develop the same disease or be treated with same drug. With the availability of GWAS data, new 'chip-based' approaches have been developed to estimate heritability from population-based samples24. However, data from at least a few thousand individuals are required to achieve an accurate estimate of heritability by these methods25. Such methods, therefore, can be applied to estimate the heritability of treatment efficacy for commonly used drugs, but not the less frequent ADRs.
In a study of GWAS data from 2,085 patients with T2DM, heritability of glycaemic response to metformin was estimated to be up to 34% (P = 0.02)23. Furthermore, this investigation also found that the heritability is probably the result of many common response variants with small to moderate effect sizes scattered across the genome23. These results suggest that the genetic architecture of metformin efficacy is similar to that of T2DM and other complex traits. This similarity between the genetic architectures of T2DM and the treatment efficacy of metformin is likely to be rooted in the multifactorial aetiology of the disease. Variants in different genes or pathways might affect metformin treatment efficacy in patients whose pathophysiology is heterogeneous (for example, those individuals who are predominantly insulin resistant or those whose insulin secretion is deficient). Similar to metformin, other antidiabetic agents are also used to treat patients with a heterogeneous pathophysiology. We, therefore, anticipate that the genetic architecture of treatment efficacy for other antidiabetic agents will be similar to that of the metformin response.
To appreciate the scale of the heritability estimate of 34% for glycaemic response to metformin, it is necessary to put it into the context of other complex traits. In a 2015 study, again using a population-based method, the heritability estimate for BMI was 27% (standard error 2.5%)26, which is considerably lower than the heritability estimates of ∼40–60% derived from traditional twin and family studies27. The discrepancy observed between the two methods could be explained by the fact that heritability is underestimated by the 'chip-based' method due to imperfect tagging and it is often overestimated by the traditional twin studies due to common environment confounding28. Therefore the actual heritability of glycaemic response to metformin could be even higher than what has been estimated from GWAS data. In addition, chip-based heritability estimates also suffer underestimation due to the incomplete coverage of contributions from rare variants, whereas traditional twin and family studies are unbiased in this regard24. Finally, the diversity of the microbiota residing in the gut might also contribute to the variable response to metformin. For example, metformin-associated changes in the gut microbiome accounts for a considerable proportion of the difference in gut taxonomic composition between patients with T2DM and control individuals without the disease29. Examining the diversity and composition of the gut microbiome might, therefore, enable the identification of novel targets for the prevention or management of T2DM as the microbiota genome is easier to modify with prebiotics or probiotics compared with the host genome30.
Notably, twin and family studies have been used to estimate the heritability of physiological response to antidiabetic agents in participants without T2DM. For example, in a twin-family study of 100 healthy twins and 25 siblings, the heritability of GLP-1 stimulated insulin secretion during hyperglycaemia was 53%31. In another family study, the heritability of tolbutamide-stimulated insulin secretion (Acute Insulin Responsetolbutamide) in 284 healthy family members of patients with T2DM was estimated to be 69%32. The results of these twin and family studies that include non-diabetic individuals have demonstrated that a large component of the variation in physiological response to antidiabetic drugs is contributed by genetic variants. However, to what extent such high heritability estimates are comparable to that of glycaemic response estimated from population-based studies of patients with T2DM is unclear. Two reasons might account for different heritability estimates between the two study designs. Firstly, twin and family studies are often performed in controlled settings, which have less environmental variance than real-world patient populations. Consequently, the same genetic effect sizes could lead to higher heritability estimates. Secondly, the pharmacodynamics in patients with T2DM might differ from that in healthy individuals, as the mechanism of glycaemic homeostasis could vary by physiological states in which different functional pathways are involved33.
Most of the robust findings in pharmacogenomic studies to date are related to severe ADRs2. The variants associated with these rare ADRs often confer a large risk. For example, the HLA-B*57:01 allele results in an 80-fold (P = 9.0 × 10−9) increased risk of flucloxacillin-induced liver injury compared with non-carriers of this allele34. Encouraged by findings like this, many of the drug-response variants have been proposed to have considerable, clinically significant effects on treatment outcome35,36. This proposal is supported by the hypothesis that drug response variants lack the evolutionary constraint that has filtered out large disease risk variants via natural selection35. However, two explanations exist as to why large effect pharmacogenomic variants are unusual, especially for variants affecting treatment efficacy. Firstly, pharmaceutical interventions often achieve clinical effect via complex metabolic networks, which rely on redundant pathways and synergistic effects to maintain their robustness when confronted with external stimuli37. Partial or complete impairment of one node in the network is, therefore, more likely to have a marginal effect on treatment efficacy than a complete shutdown of all relevant pathways. Secondly, the established spectrum of large effect ADR variants might also reflect a publication bias, which accumulated the so-called 'low hanging fruit' that have been identified by pharmacogenomic studies often using fewer than 1000 cases3,38. Although the genetic architecture of rare ADRs might be akin to those of polygenic diseases in which large effect variants dominate3, we anticipate the genetic architecture of treatment efficacy and mild ADRs would both encompass a spectrum of rare-to-common variants with moderate effect sizes. This notion is in line with the fact that rare variants with moderate effect have been successfully identified for common diseases such as T2DM by sequencing and imputation-based rare variant association studies of >100,000 samples9. Assembling large cohorts would, therefore, enable the identification of more drug response variants by future pharmacogenomic studies.
Pharmacogenomics of T2DM drugs
Owing to the considerable variability in response to existing drugs to treat diabetes mellitus, a large number of pharmacogenetic studies have been published, but only one pharmacogenomic GWAS study of metformin treatment efficacy reported39. These studies each focused on a single oral agent and have been the subject of many previous reviews40,41,42. No report exists on the pharmacogenetics of drug–drug interactions, despite a large number of patients requiring multiple agents to combat diabetes mellitus progression and for maintaining glycaemic control. In this section we summarize the replicated findings in studies of treatment efficacy and place more emphasis on the investigation of adverse effects (Table 2).
Treatment efficacy
Very few robust pharmacogenetic findings related to treatment efficacy of diabetes mellitus drugs have been reported. Previous candidate gene studies largely focused on drug transporters or metabolizing enzyme variants that have been implicated in the pharmacokinetics of drug exposure41. Variation in metformin pharmacokinetics is mainly the result of variants in the transporters SLC22A1 (which encodes solute carrier family 22 member 1, commonly known as OCT1) and SLC47A1 (which encodes MATE-1)43,44. However, the most investigated reduced-function OCT1 variants with low transporter activity had no consistent effect on glycaemic control in patients45,46,47,48. Sulfonylureas are mainly metabolized by Cytochrome P450 2C9 which is encoded by CYP2C9. Individuals with loss-of-function variants in CYP2C9 have higher drug exposure, and this in turn leads to consistent observations of greater glycaemic response than in those carrying wild-type alleles49,50,51.
Studies of potential pharmacodynamic variants, which might affect how antidiabetic agents alter glucose levels, have largely focused on the genes involved in glucose metabolism and the risk of developing T2DM. One replicated finding is seen for TCF7L2 and sulfonylurea response, where the allele associated with reduced β-cell function and, therefore, increased risk of T2DM, is also associated with reduced efficacy of sulfonylureas52,53,54. The K allele of the E23K variant in KCNJ11, which encodes the known targets of sulfonylureas, is also associated with an increased glycaemic response in multiple studies55,56,57. These observations of E23K carriers are consistent with evidence that neonates with monogenic diabetes mellitus who have causal variants in KCNJ11 could be effectively treated with sulfonylureas18. Another replicated finding is the association between the PPARG Pro12Ala variant and response to thiazolidinediones (commonly known as TZDs). A few studies, each with fewer than 200 participants, have consistently reported that the T2DM risk Pro allele is associated with poor glycaemic response58,59,60. For the newer agents such as gliptins and SGLT-2 inhibitors, pharmacogenetic studies of treatment efficacy have been sparse with only one relatively large study reporting the association and replication between rs7202877 near CTRB1/2 and response to gliptins61.
The only GWAS of any antidiabetic agent published to date identified a variant rs11212617 near the ATM locus as being associated with glycaemic response to metformin39. Independent replications were later reported in multiple cohorts of different ancestries, strengthening evidence that this variant is the most established drug response variant for an antidiabetic drug62,63. Given the moderate effect on glycaemic response to metformin, this variant is not a useful biomarker that can substantially increase our ability to accurately predict the treatment outcome in individual patients. However, as this variant has no functional link to any known metformin mode-of-action, it demonstrates that such a pharmacogenomic discovery can reveal novel mechanisms of action of an antidiabetic drug, which might in turn identify further pathways to target with new therapeutic agents. The finding that variants near ATM are associated with metformin response has prompted further study of the genes at this locus in relation to glucose metabolism and metformin response. For example, in a small study of patients with ataxia telangiectasia who have recessive loss-of-function mutations in ATM, the investigators identified impaired glucose tolerance and insulin resistance, which supports a potential role for ATM in glucose metabolism and the response to metformin64.
ADRs
Pharmacogenetic studies have also been performed to help in the understanding of ADRs associated with antidiabetic drugs. Key ADRs studied include sulfonylurea-induced hypoglycaemia, TZD-associated oedema, hepatotoxicity, heart failure and metformin-associated gastrointestinal disturbance65,66,67,68,69. To date, no studies have been published on the pharmacogenomics of the potentially fatal but rare ADRs of metformin-associated lactic acidosis70. Thus far, the investigators in all the published pharmacogenetic studies have adopted a candidate gene approach. For sulfonylurea-induced hypoglycaemia, a number of small studies using no more than 108 patients have been conducted and they consistently showed that loss-of-function CYP2C9 variants, which are associated with increased drug exposure66, are also associated with a higher risk of hypoglycaemia65, which is consistent with the efficacy studies showing that these variants are associated with greater glucose reduction than wild-type alleles45. For TZDs, the results have indicated that variants in GSTT1 and CYP2C19 are associated with troglitazone-induced hepatotoxicity68,71; while variants in NFATC2 are associated with the rate of rosiglitazone-induced oedema67. Although some safety concerns have been raised about gliptins and SGLT-2 inhibitors, such as genital and urinary tract infections associated with SGLT-2 inhibitors and the pancreatitis and liver dysfunction associated with gliptins70, to our knowledge, no pharmacogenetic reports on these ADRs exist.
Metformin causes gastrointestinal disturbance in as many as 20–40% of patients with 5–10% of patients not being able to tolerate the drug70. The biological mechanism underlying gastrointestinal intolerance to metformin remains poorly understood. On the basis of the hypothesis that individuals who are intolerant to metformin are exposed to higher concentrations of the drug in the gastrointestinal tract, investigators have explored variants in transporters such as OCT1 (Ref. 72). In a large study of 2,166 patients, both reduced-function variants in OCT1 and co-medication of certain OCT1 inhibitors (that is, verapamil, proton pump inhibitors, citalopram, codeine and doxazosin) significantly increased the risk of metformin-induced gastrointestinal side effects69. The combination of carrying the genetic variants and taking co-medications could result in a fourfold (P <0.001) increased risk of gastrointestinal side effects69. Therefore this large effect on metformin-induced gastrointestinal side effects by these OCT1 variants have the potential to be translated into clinical practice by prescreening the patients for OCT1 genotype and co-medication of OCT1 inhibitors. Moreover, this study highlighted two key factors for successful pharmacogenomics of antidiabetic drugs: large sample sizes are required for adequate statistical power; and the need to consider co-prescribed medication that potentially interacts with these agents69. This result has since been replicated in an independent study in which the same genetic variants were associated with an increased risk (OR 2.3; P = 0.02) of less severe metformin intolerance73.
Genetics and drug target validation
A major challenge in the modern pharmaceutical industry is that <15% of drugs entering the development pipeline make it to market74, a vastly expensive, inefficient and wasteful process. Toxicity and lack of efficacy contribute to the high failure rate of new drugs in development, which reflects the ineffectiveness of conventional target validation methods used in preclinical studies. Moreover, the model systems used in the preclinical studies often fail to represent real biological systems working in humans.
Genetic variants that arise as a result of historical mutation and recombination events can be viewed as naturally occurring experiments that perturb human gene function. These naturally occurring mutations are an opportunity to see the effect of perturbing a gene (for example, by a potential novel drug) on disease risk and off-target effects without the need to develop and trial the drug. For example, in a large survey of 61,104 drugs across the various stages of development, those candidates that targeted proteins encoded by genes with robust human genetic evidence (that is evidence from GWAS or an association with diseases in OMIM) are twice as likely to be therapeutically valid75.
To address the question of how to harness human genetic evidence to guide target validation in drug development, the 'therapeutic hypothesis' has been proposed76. Central to this approach is the concept of a genetic evidence based 'dose-response' curve. An example of translating such a hypothesis into a new drug is the development of the PCSK9 inhibitors to reduce levels of LDL cholesterol77. Rare 'gain-of-function' variants in PSCK9 lead to high levels of LDL cholesterol and an increased risk of coronary heart disease78. Conversely, rare 'loss-of-function' variants result in lower LDL cholesterol levels and a reduced risk of coronary heart disease79. GWAS also identified common variants in other genes such as SORT1 and LDLR with mild effect on LDL cholesterol levels and risk of coronary heart disease80,81. These experiments (which are essentially designed by nature) have demonstrated that reducing levels of LDL cholesterol and the risk of coronary heart disease is possible by inhibiting the function of PCSK9 without any observable adverse effects77. With further support by other mechanistic evidence derived from model systems, a new generation of LDL-cholesterol-lowering PCSK9 inhibitors have been developed, tested and licensed for use82.
Dozens of common and rare variants have been convincingly associated with glycaemic control and the risk of developing T2DM10. Representatives from both academia and pharmaceutical companies have now formed the Accelerating Medicines Partnership to enhance the translation of human genomic research outputs into the development of new drugs (Accelerating Medicines Partnership)83. As one of the three prioritized areas, the Accelerating Medicines Partnership aims to establish an open-access portal for T2DM genetics research (http://www.type2diabetesgenetics.org), which will pool genomic and phenotyping data to facilitate novel data mining efforts83. One target of potential interest raised by the Accelerating Medicines Partnership is the zinc-transporter-encoding SLC30A8. The use of GWAS has established robust evidence that the common coding variant Arg325Trp is associated with the risk of T2DM84. The results of mechanistic studies in humans and mice have both indicated that the reduced zinc transporter activity allele is associated with an increased risk of T2DM85,86. However, in a sequencing-based study of ∼150,000 individuals, rare protein-truncating variants in SLC30A8 protected the carriers from developing T2DM9. This conflicting genetic evidence does not provide a consistent dose-response curve based on the functional characterization of the variants87. Further mechanistic studies, especially those involving intensive phenotyping of individuals carrying rare extreme functional SLC30A8 variants will be useful to validate whether inhibiting or enhancing the zinc transporter 8 function could be the therapeutic option for treating T2DM. The example provided by SLC30A8 demonstrates that human genetic evidence is not always sufficient to validate a gene or protein as a drug target, especially in situations when uncertainty surrounds the exact mechanism of how target genes alter a phenotype of interest.
The discovery of SGLT-2 inhibitors to treat T2DM is another example of how genetic evidence can be used to assist drug development (Fig. 2). Early evidence indicated that phlorizin, a natural inhibitor of SGLT-2 and isolated from the bark of apple trees, restored euglycaemia and insulin sensitivity in animal models of T2DM88. After the cloning of SLC5A2 which encodes SGLT-2, functional variants in SGLT-2 have been linked to familial renal glycosuria (OMIM)89. Patients with familial renal glycosuria, who carry a spectrum of >50 mild heterozygote to severe homozygote SGLT-2 loss-of-function variants, have different levels of glycosuria but apparently normal renal functions, normal glucose concentrations and generally good health90. Here the genetic evidence not only validated the therapeutic potential of inhibiting SGLT-2 but also provided critical evidence that selective inhibitors targeting the protein would result in no long-term ADRs. Consequently, several selective SGLT-2 inhibitors have been successfully developed and licensed to treat T2DM14. Aside from their glucose-lowering effect, these SGLT-2 inhibitors have an average 1.63 kg (P <0.001) weight reduction benefit16,91, which is increasingly considered an important component in the management of T2DM92.

The y axis represents a range of glucose levels, in which the high range represents the hyperglycaemic state observed in type 2 diabetes mellitus as compared to the normal range observed in healthy individuals or those patients with familial renal glycosuria. The x axis represents a spectrum of naturally occurring sodium–glucose cotransporter 2 (SGLT-2) loss-of-function variants observed in patients with familial renal glycosuria. The variants were ordered from the mild heterozygotes to severe homozygotes as defined by the resulting severity of glycosuria. For on-target adverse reactions, the benign glucosuria and apparently normal health observed in these patients supports the safety profile of selectively inhibiting SGLT-2 function in a wide dose window.
Pharmacogenomics and drug discovery
Conventional pharmacogenetics, which is based on candidate genes, has been used in all stages of the drug development pipeline, from target identification to clinical trials and postmarket analysis of ADRs35. As pharmacogenetics moves to pharmacogenomics, this genome-wide-hypothesis-free approach could also have more applications in drug discovery and development20.
The proposed 'therapeutic hypothesis' is based on the principle that human genetics is an experiment of nature, which shows what phenotypic outcomes the natural perturbations of gene functions can lead to in a human population76. Clearly human genetics has the advantage that the experimental system is in humans as opposed to cell or animal model systems used in preclinical studies. However, the ideal system for validating a therapeutic hypothesis would be a genetic study carried out in the exact context the new drug is developed for. Genetic evidence of disease risk could be useful in validating the targets for a preventive drug but less informative for predicting therapeutic potential for disease management. For example, let us consider using human genetic evidence to validate candidate drug targets to improve glycaemic control in patients with T2DM. The most available candidates would be those genes with established variants associated with the risk of developing T2DM or poor glycaemic control in the general population. However, the mechanism regulating glycaemia has been suggested to vary between different physiological states33. The variants affecting glycaemia in normal individuals and those associated with T2DM risk only partially overlap33, which suggests perturbations of glycaemia are not always linked to the risk of T2DM. Similarly, the variants conferring risk to T2DM have little effect on the rate of disease progression93, which suggests that the mechanisms controlling glycaemia when T2DM has developed might be different from those involved in its development. Consequently, targeting genes involved in the regulation of glycaemia in healthy individuals or the risk of T2DM might not have the desired glycaemic effect on the management of T2DM, especially if their biological functions have been altered by the onset of the disease. In the context of disease management, pharmacogenomic studies of existing antidiabetic drugs can identify genes involved in glycaemic control in patients with T2DM. Robust pharmacogenomic findings would, therefore, provide physiological-state-specific information for drug target validation in addition to other evidence of normal glycaemic control and risk of T2DM derived from human genetic studies.
Table 1 outlines the differences between pharmacogenomic studies of drugs used to treat T2DM and genomics of other diabetes mellitus-related traits. Compared to the wealth of findings from other areas of human genetics, to date pharmacogenomics has yielded less robust results, in part owing to the challenge to assemble tens of thousands of samples that could provide adequate statistical power to detect variants with moderate effect sizes. However, we anticipate future drug discovery and drug development to benefit more from the pharmacogenomic findings derived from larger samples that are increasingly available from biobanks linked to electronic health record data (for example the UKBiobank). Notably, longitudinal data from clinical trials or health record linkage enable the study of outcomes other than simple measures of glycaemia. For example, given a sufficient sample size, using pharmacogenomics to assess cardiovascular endpoints of antidiabetic drugs might be possible. In addition, large samples would also enable pharmacogenomics to jointly analyse multiple outcomes (such as, both glycaemic benefit and weight benefit of metformin), which would help to identify variants associated with pleiotropic effects of antidiabetic drugs.
Avoiding ADRs or off-target effects is also an important consideration in drug development. The very existence of ADRs is an illustration of our incomplete understanding of the complex interactions between our biological system and any designed interventions94. The increased availability of genome-wide screening tools will enable the identification of more ADR variants at an early stage of drug development, which has a number of clear advantages. For example, if ADR variants are identified before phase III clinical trials, recruitment-by-genotype trials could be performed to evaluate the efficacy and risk in stratified patient subgroups35. When a drug is approved for marketing, understanding the genetic basis of severe ADRs could also help the continuous development of the drug by prescreening the genotype ahead of treatment95.
A systems biology view of drug response
One of the biggest challenges facing contemporary biological research is to understand the complex biological networks that function in a living system96. This complexity can be seen in T2DM, where the genes harbouring the established aetiological variants have an excess of interactions within a high-confidence interaction network97. The variants in this network affect the protein function, stability of protein or transcript, and expression of individual genes, which collectively perturb or rewire the network to alter the risk of T2DM98,99. Interestingly, when considering a network of disease genes and genes encoding known drug targets, the distance between genes reported in GWAS and known drug targets are shorter than those between random gene–drug targets pairs100. This is predominantly driven by a threefold enrichment of drug target genes among the first neighbours of the GWAS reported genes. More evidence of direct overlap between the drug target genes and disease risk genes was also presented in a gene set enrichment analysis101. In this study, a set of 102 target genes for existing diabetes mellitus drugs (for example, insulin, metformin and TZDs), as curated from the literature, showed significant enrichment of genetic association with T2DM susceptibility101. Such findings indicate that potential drug targets are enriched in a disease network, but might only be identified when considering the network as a whole, rather than individual disease genes.
When modelling a drug intervention in a functional network, it is important to consider how the drug alters the functional network at the level of the cell, the tissue and the disease state, which will ultimately determine the beneficial and harmful effects of the drug. To date, our knowledge of biological networks has been largely limited to the generic, static models lacking such contextual information102. Research in systems biology is beginning to offer more comprehensive cell-lineage and tissue-specific networks103,104,105, which will enable modelling of drug interventions in specific contexts such as the cell type, the tissue and the physiological state. Such analyses will provide insights into how drugs can achieve the desired therapeutic effect in target tissues or known sites of action, but importantly highlight the potential undesirable off-target effects in other contexts. Moreover, adopting a systems-wide approach might change the focus of drug development for complex diseases such as T2DM from targeting an individual protein or gene to systems-wide attacks on multiple dynamic targets106.
Conclusions
The availability of affordable high-throughput genomic technologies has expanded our knowledge about the multifactorial aetiology of T2DM. Studies adopting such genome-wide approaches to investigate the response of existing antidiabetic drugs have been limited, but have the potential to improve our understanding of the biological mechanisms underlying treatment efficacy and adverse effects. With major investments in precision medicine in the USA107, the 100,000 genomes project in the UK108, and the EU-funded stratified medicine in diabetes mellitus initiative Innovative Medicines Initiative: DIabetes REsearCh on patient straTification (known as IMI-DIRECT)109, more findings from adequately powered pharmacogenomic studies are expected to complement other human genetic discoveries to facilitate more efficient antidiabetic drug discovery programs.
References
Visscher, P. M., Brown, M. A., McCarthy, M. I. & Yang, J. Five years of GWAS discovery. Am. J. Hum. Genet. 90, 7–24 (2012).
Daly, A. K. Pharmacogenomics of adverse drug reactions. Genome Med. 5, 5 (2013).
Zhou, K. & Pearson, E. R. Insights from genome-wide association studies of drug response. Annu. Rev. Pharmacol. Toxicol. 53, 299–310 (2013).
American Diabetes, A. Diagnosis and classification of diabetes mellitus. Diabetes Care 27, S5–S10 (2004).
Stumvoll, M., Goldstein, B. J. & van Haeften, T. W. Type 2 diabetes: principles of pathogenesis and therapy. Lancet 365, 1333–1346 (2005).
Morris, A. P. et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat. Genet. 44, 981–990 (2012).
Prasad, R. B. & Groop, L. Genetics of type 2 diabetes — pitfalls and possibilities. Genes (Basel) 6, 87–123 (2015).
Steinthorsdottir, V. et al. Identification of low-frequency and rare sequence variants associated with elevated or reduced risk of type 2 diabetes. Nat. Genet. 46, 294–298 (2014).
Flannick, J. et al. Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat. Genet. 46, 357–363 (2014).
Mohlke, K. L. & Boehnke, M. Recent advances in understanding the genetic architecture of type 2 diabetes. Hum. Mol. Genet. 24, R85–92 (2015).
Holman, R. R., Paul, S. K., Bethel, M. A., Matthews, D. R. & Neil, H. A. 10-year follow-up of intensive glucose control in type 2 diabetes. N. Engl. J. Med. 359, 1577–1589 (2008).
Stratton, I. M. et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ 321, 405–412 (2000).
[No authors listed.] Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 352, 837–853 (1998).
Bailey, C. J. The current drug treatment landscape for diabetes and perspectives for the future. Clin. Pharmacol. Ther. 98, 170–184 (2015).
Deacon, C. F. Dipeptidyl peptidase-4 inhibitors in the treatment of type 2 diabetes: a comparative review. Diabetes Obes. Metab. 13, 7–18 (2011).
Tahrani, A. A., Barnett, A. H. & Bailey, C. J. SGLT inhibitors in management of diabetes. Lancet Diabetes Endocrinol. 1, 140–151 (2013).
Pearson, E. R. et al. Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet 362, 1275–1281 (2003).
Gloyn, A. L. et al. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N. Engl. J. Med. 350, 1838–1849 (2004).
Carr, D. F., Alfirevic, A. & Pirmohamed, M. Pharmacogenomics: current state-of-the-art. Genes (Basel) 5, 430–443 (2014).
Goldstein, D. B., Tate, S. K. & Sisodiya, S. M. Pharmacogenetics goes genomic. Nat. Rev. Genet. 4, 937–947 (2003).
Goldstein, D. B. et al. Sequencing studies in human genetics: design and interpretation. Nat. Rev. Genet. 14, 460–470 (2013).
Mizzi, C. et al. Personalized pharmacogenomics profiling using whole-genome sequencing. Pharmacogenomics 15, 1223–1234 (2014).
Zhou, K. et al. Heritability of variation in glycaemic response to metformin: a genome-wide complex trait analysis. Lancet Diabetes Endocrinol. 2, 481–487 (2014).
Yang, J. et al. Genome partitioning of genetic variation for complex traits using common SNPs. Nat. Genet. 43, 519–525 (2011).
Visscher, P. M. et al. Statistical power to detect genetic (co)variance of complex traits using SNP data in unrelated samples. PLoS Genet. 10, e1004269 (2014).
Yang, J. et al. Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index. Nat. Genet. 47, 1114–1120 (2015).
Elks, C. E. et al. Variability in the heritability of body mass index: a systematic review and meta-regression. Front. Endocrinol. (Lausanne) 3, 29 (2012).
Yang, J. et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 42, 565–569 (2010).
Forslund, K. et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 528, 262–266 (2015).
McCreight, L. J., Bailey, C. J. & Pearson, E. R. Metformin and the gastrointestinal tract. Diabetologia 59, 426–435 (2016).
Simonis-Bik, A. M. et al. Genetic influences on the insulin response of the beta cell to different secretagogues. Diabetologia 52, 2570–2577 (2009).
Gjesing, A. P. et al. High heritability and genetic correlation of intravenous glucose- and tolbutamide-induced insulin secretion among non-diabetic family members of type 2 diabetic patients. Diabetologia 57, 1173–1181 (2014).
De Silva, N. M. & Frayling, T. M. Novel biological insights emerging from genetic studies of type 2 diabetes and related metabolic traits. Curr. Opin. Lipidol. 21, 44–50 (2010).
Daly, A. K. et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat. Genet. 41, 816–819 (2009).
Harper, A. R. & Topol, E. J. Pharmacogenomics in clinical practice and drug development. Nat. Biotechnol. 30, 1117–1124 (2012).
Maranville, J. C. & Cox, N. J. Pharmacogenomic variants have larger effect sizes than genetic variants associated with other dichotomous complex traits. Pharmacogenomics J. http://dx.doi.org/10.1038/tpj.2015.47 (2015).
Hellerstein, M. K. Exploiting complexity and the robustness of network architecture for drug discovery. J. Pharmacol. Exp. Ther. 325, 1–9 (2008).
Nelson, M. R. et al. Genome-wide approaches to identify pharmacogenetic contributions to adverse drug reactions. Pharmacogenomics J. 9, 23–33 (2009).
Zhou, K. et al. Common variants near ATM are associated with glycemic response to metformin in type 2 diabetes. Nat. Genet. 43, 117–120 (2011).
Becker, M. L., Pearson, E. R. & Tkac, I. Pharmacogenetics of oral antidiabetic drugs. Int. J. Endocrinol. 2013, 686315 (2013).
Semiz, S., Dujic, T. & Causevic, A. Pharmacogenetics and personalized treatment of type 2 diabetes. Biochem. Med. (Zagreb) 23, 154–171 (2013).
Todd, J. N. & Florez, J. C. An update on the pharmacogenomics of metformin: progress, problems and potential. Pharmacogenomics 15, 529–539 (2014).
Shu, Y. et al. Effect of genetic variation in the organic cation transporter 1, OCT1, on metformin pharmacokinetics. Clin. Pharmacol. Ther. 83, 273–280 (2008).
Graham, G. G. et al. Clinical pharmacokinetics of metformin. Clin. Pharmacokinet. 50, 81–98 (2011).
Zhou, K. et al. Reduced-function SLC22A1 polymorphisms encoding organic cation transporter 1 and glycemic response to metformin: a GoDARTS study. Diabetes 58, 1434–1439 (2009).
Jablonski, K. A. et al. Common variants in 40 genes assessed for diabetes incidence and response to metformin and lifestyle intervention in the diabetes prevention program. Diabetes 59, 2672–2681 (2010).
Becker, M. L. et al. Genetic variation in the organic cation transporter 1 is associated with metformin response in patients with diabetes mellitus. Pharmacogenomics J. 9, 242–247 (2009).
Shu, Y. et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J. Clin. Invest. 117, 1422–1431 (2007).
Zhou, K. et al. Loss-of-function CYP2C9 variants improve therapeutic response to sulfonylureas in type 2 diabetes: a Go-DARTS study. Clin. Pharmacol. Ther. 87, 52–56 (2010).
Becker, M. L. et al. Cytochrome P450 2C9 *2 and *3 polymorphisms and the dose and effect of sulfonylurea in type II diabetes mellitus. Clin. Pharmacol. Ther. 83, 288–292 (2008).
Suzuki, K. et al. Effect of CYP2C9 genetic polymorphisms on the efficacy and pharmacokinetics of glimepiride in subjects with type 2 diabetes. Diabetes Res. Clin. Pract. 72, 148–154 (2006).
Pearson, E. R. et al. Variation in TCF7L2 influences therapeutic response to sulfonylureas: a GoDARTs study. Diabetes 56, 2178–2182 (2007).
Schroner, Z. et al. Effect of sulphonylurea treatment on glycaemic control is related to TCF7L2 genotype in patients with type 2 diabetes. Diabetes Obes. Metab. 13, 89–91 (2011).
Javorsky, M. et al. Association between TCF7L2 genotype and glycemic control in diabetic patients treated with gliclazide. Int. J. Endocrinol. 2013, 374858 (2013).
Javorsky, M. et al. KCNJ11 gene E23K variant and therapeutic response to sulfonylureas. Eur. J. Intern. Med. 23, 245–249 (2012).
Feng, Y. et al. Ser1369Ala variant in sulfonylurea receptor gene ABCC8 is associated with antidiabetic efficacy of gliclazide in Chinese type 2 diabetic patients. Diabetes Care 31, 1939–1944 (2008).
Zhang, H., Liu, X., Kuang, H., Yi, R. & Xing, H. Association of sulfonylurea receptor 1 genotype with therapeutic response to gliclazide in type 2 diabetes. Diabetes Res. Clin. Pract. 77, 58–61 (2007).
Pei, Q. et al. PPAR-γ2 and PTPRD gene polymorphisms influence type 2 diabetes patients' response to pioglitazone in China. Acta Pharmacol. Sin. 34, 255–261 (2013).
Kang, E. S. et al. Effects of Pro12Ala polymorphism of peroxisome proliferator-activated receptor γ2 gene on rosiglitazone response in type 2 diabetes. Clin. Pharmacol. Ther. 78, 202–208 (2005).
Hsieh, M. C. et al. Common polymorphisms of the peroxisome proliferator-activated receptor-γ (Pro12Ala) and peroxisome proliferator-activated receptor-γ coactivator-1 (Gly482Ser) and the response to pioglitazone in Chinese patients with type 2 diabetes mellitus. Metabolism 59, 1139–1144 (2010).
t Hart, L. M. et al. The CTRB1/2 locus affects diabetes susceptibility and treatment via the incretin pathway. Diabetes 62, 3275–3281 (2013).
van Leeuwen, N. et al. A gene variant near ATM is significantly associated with metformin treatment response in type 2 diabetes: a replication and meta-analysis of five cohorts. Diabetologia 55, 1971–1977 (2012).
Zhou, Y. et al. RS11212617 is associated with metformin treatment response in type 2 diabetes in Shanghai local Chinese population. Int. J. Clin. Pract. 68, 1462–1466 (2014).
Connelly, P. J. et al. Recessive mutations in the cancer gene Ataxia Telangiectasia Mutated (ATM), at a locus previously associated with metformin response, cause dysglycaemia and insulin resistance. Diabet. Med. 33, 371–375 (2016).
Gokalp, O. et al. Mild hypoglycaemic attacks induced by sulphonylureas related to CYP2C9, CYP2C19 and CYP2C8 polymorphisms in routine clinical setting. Eur. J. Clin. Pharmacol. 67, 1223–1229 (2011).
Zhang, Y. et al. Influence of CYP2C9 and CYP2C19 genetic polymorphisms on pharmacokinetics of gliclazide MR in Chinese subjects. Br. J. Clin. Pharmacol. 64, 67–74 (2007).
Bailey, S. D. et al. Variation at the NFATC2 locus increases the risk of thiazolidinedione-induced edema in the Diabetes REduction Assessment with ramipril and rosiglitazone Medication (DREAM) study. Diabetes Care 33, 2250–2253 (2010).
Watanabe, I. et al. A study to survey susceptible genetic factors responsible for troglitazone-associated hepatotoxicity in Japanese patients with type 2 diabetes mellitus. Clin. Pharmacol. Ther. 73, 435–455 (2003).
Dujic, T. et al. Association of organic cation transporter 1 with intolerance to metformin in type 2 diabetes: a GoDARTS study. Diabetes 64, 1786–1793 (2015).
Bailey, C. J. Safety of antidiabetes medications: an update. Clin. Pharmacol. Ther. 98, 185–195 (2015).
Kumashiro, R. et al. Association of troglitazone-induced liver injury with mutation of the cytochrome P450 2C19 gene. Hepatol. Res. 26, 337–342 (2003).
Tarasova, L. et al. Association of genetic variation in the organic cation transporters OCT1, OCT2 and multidrug and toxin extrusion 1 transporter protein genes with the gastrointestinal side effects and lower BMI in metformin-treated type 2 diabetes patients. Pharmacogenet. Genomics 22, 659–666 (2012).
Dujic, T. et al. Organic cation transporter 1 variants and gastrointestinal side effects of metformin in patients with type 2 diabetes. Diabet. Med. 33, 511–514 (2016).
Hay, M., Thomas, D. W., Craighead, J. L., Economides, C. & Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 32, 40–51 (2014).
Nelson, M. R. et al. The support of human genetic evidence for approved drug indications. Nat. Genet. 47, 856–860 (2015).
Plenge, R. M., Scolnick, E. M. & Altshuler, D. Validating therapeutic targets through human genetics. Nat. Rev. Drug Discov. 12, 581–594 (2013).
Cohen, J. C. Emerging LDL therapies: using human genetics to discover new therapeutic targets for plasma lipids. J. Clin. Lipidol. 7, S1–5 (2013).
Abifadel, M. et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 34, 154–156 (2003).
Cohen, J. C., Boerwinkle, E., Mosley, T. H. Jr & Hobbs, H. H. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 354, 1264–1272 (2006).
Global Lipids Genetics, C. et al. Discovery and refinement of loci associated with lipid levels. Nat. Genet. 45, 1274–1283 (2013).
Schunkert, H. et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat. Genet. 43, 333–338 (2011).
Stein, E. A. et al. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N. Engl. J. Med. 366, 1108–1118 (2012).
Mullard, A. Drug makers and NIH team up to find and validate targets. Nat. Rev. Drug Discov. 13, 241–243 (2014).
Lyssenko, V. et al. Clinical risk factors, DNA variants, and the development of type 2 diabetes. N. Engl. J. Med. 359, 2220–2232 (2008).
Tamaki, M. et al. The diabetes-susceptible gene SLC30A8/ZnT8 regulates hepatic insulin clearance. J. Clin. Invest. 123, 4513–4524 (2013).
Nicolson, T. J. et al. Insulin storage and glucose homeostasis in mice null for the granule zinc transporter ZnT8 and studies of the type 2 diabetes-associated variants. Diabetes 58, 2070–2083 (2009).
Pearson, E. Zinc transport and diabetes risk. Nat. Genet. 46, 323–324 (2014).
Rossetti, L., Smith, D., Shulman, G. I., Papachristou, D. & DeFronzo, R. A. Correction of hyperglycemia with phlorizin normalizes tissue sensitivity to insulin in diabetic rats. J. Clin. Invest. 79, 1510–1515 (1987).
Santer, R. & Calado, J. Familial renal glucosuria and SGLT2: from a mendelian trait to a therapeutic target. Clin. J. Am. Soc. Nephrol. 5, 133–141 (2010).
Sabino-Silva, R. et al. The Na+/glucose cotransporters: from genes to therapy. Braz. J. Med. Biol. Res. 43, 1019–1026 (2010).
Zhang, M. et al. Dapagliflozin treatment for type 2 diabetes: a systematic review and meta-analysis of randomized controlled trials. Diabetes Metab. Res. Rev. 30, 204–221 (2014).
Van Gaal, L. & Scheen, A. Weight management in type 2 diabetes: current and emerging approaches to treatment. Diabetes Care 38, 1161–1172 (2015).
Zhou, K. et al. Clinical and genetic determinants of progression of type 2 diabetes: a DIRECT study. Diabetes Care 37, 718–724 (2014).
Chang, R. L., Xie, L., Xie, L., Bourne, P. E. & Palsson, B. O. Drug off-target effects predicted using structural analysis in the context of a metabolic network model. PLoS Comput. Biol. 6, e1000938 (2010).
Mallal, S. et al. HLA-B*5701 screening for hypersensitivity to abacavir. N. Engl. J. Med. 358, 568–579 (2008).
Csermely, P., Korcsmaros, T., Kiss, H. J., London, G. & Nussinov, R. Structure and dynamics of molecular networks: a novel paradigm of drug discovery: a comprehensive review. Pharmacol. Ther. 138, 333–408 (2013).
Taneera, J. et al. A systems genetics approach identifies genes and pathways for type 2 diabetes in human islets. Cell Metab. 16, 122–134 (2012).
Gaulton, K. J. et al. Genetic fine mapping and genomic annotation defines causal mechanisms at type 2 diabetes susceptibility loci. Nat. Genet. 47, 1415–1425 (2015).
Carter, H., Hofree, M. & Ideker, T. Genotype to phenotype via network analysis. Curr. Opin. Genet. Dev. 23, 611–621 (2013).
Cao, C. & Moult, J. GWAS and drug targets. BMC Genomics 15, S5 (2014).
Segre, A. V., Wei, N., Altshuler, D. & Florez, J. C. Pathways targeted by antidiabetes drugs are enriched for multiple genes associated with type 2 diabetes risk. Diabetes 64, 1470–1483 (2015).
Ideker, T. & Krogan, N. J. Differential network biology. Mol. Syst. Biol. 8, 565 (2012).
Greene, C. S. et al. Understanding multicellular function and disease with human tissue-specific networks. Nat. Genet. 47, 569–576 (2015).
Bossi, A. & Lehner, B. Tissue specificity and the human protein interaction network. Mol. Syst. Biol. 5, 260 (2009).
Magger, O., Waldman, Y. Y., Ruppin, E. & Sharan, R. Enhancing the prioritization of disease-causing genes through tissue specific protein interaction networks. PLoS Comput. Biol. 8, e1002690 (2012).
Erler, J. T. & Linding, R. Network medicine strikes a blow against breast cancer. Cell 149, 731–733 (2012).
Collins, F. S. & Varmus, H. A new initiative on precision medicine. N. Engl. J. Med. 372, 793–795 (2015).
Siva, N. UK gears up to decode 100,000 genomes from NHS patients. Lancet 385, 103–104 (2015).
Koivula, R. W. et al. Discovery of biomarkers for glycaemic deterioration before and after the onset of type 2 diabetes: rationale and design of the epidemiological studies within the IMI DIRECT Consortium. Diabetologia 57, 1132–1142 (2014).
Miller, R. A. et al. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 494, 256–260 (2013).
Mulherin, A. J. et al. Mechanisms underlying metformin-induced secretion of glucagon-like peptide-1 from the intestinal L cell. Endocrinology 152, 4610–4619 (2011).
Gribble, F. M. & Reimann, F. Pharmacological modulation of KATP channels. Biochem. Soc. Trans. 30, 333–339 (2002).
Dornhorst, A. Insulinotropic meglitinide analogues. Lancet 358, 1709–1716 (2001).
Yki-Jarvinen, H. Thiazolidinediones. N. Engl. J. Med. 351, 1106–1118 (2004).
Verspohl, E. J. Novel therapeutics for type 2 diabetes: incretin hormone mimetics (glucagon-like peptide-1 receptor agonists) and dipeptidyl peptidase-4 inhibitors. Pharmacol. Ther. 124, 113–138 (2009).
Bailey, C. J. Renal glucose reabsorption inhibitors to treat diabetes. Trends Pharmacol. Sci. 32, 63–71 (2011).
Acknowledgements
E.R.P. holds a Wellcome Trust New Investigator Award 102820/Z/13/Z.
Author information
Authors and Affiliations
Contributions
All authors contributed to all aspects of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Related links
Rights and permissions
About this article

Cite this article
Zhou, K., Pedersen, H., Dawed, A. et al. Pharmacogenomics in diabetes mellitus: insights into drug action and drug discovery. Nat Rev Endocrinol 12, 337–346 (2016). https://doi.org/10.1038/nrendo.2016.51
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrendo.2016.51
This article is cited by
-
MIR4532 gene variant rs60432575 influences the expression of KCNJ11 and the sulfonylureas-stimulated insulin secretion
Endocrine (2019)
-
MicroRNA profiling of diabetic atherosclerosis in a rat model
European Journal of Medical Research (2018)
-
Actions of metformin and statins on lipid and glucose metabolism and possible benefit of combination therapy
Cardiovascular Diabetology (2018)
-
Interdisciplinary approach to compensation of hypoglycemia in diabetic patients with chronic heart failure
Heart Failure Reviews (2018)
-
Glucose uptake of the muscle and adipose tissues in diabetes and obesity disease models: evaluation of insulin and β3-adrenergic receptor agonist effects by 18F-FDG
Annals of Nuclear Medicine (2017)
