
Abstract
Self-monitoring of blood glucose (SMBG) is a core component of diabetes management. However, the International Diabetes Federation recommends that SMBG be performed in a structured manner and that the data are accurately interpreted and used to take appropriate therapeutic actions. We designed a study to evaluate the impact of structured SMBG on glycemic control in non-insulin-treated type 2 diabetes (T2DM) patients. The Prospective, Randomized Trial on Intensive SMBG Management Added Value in Non-insulin-Treated T2DM Patients (PRISMA) is a 12-month, prospective, multicenter, open, parallel group, randomized, and controlled trial to evaluate the added value of an intensive, structured SMBG regimen in T2DM patients treated with oral agents and/or diet. One thousand patients (500 per arm) will be enrolled at 39 clinical sites in Italy. Eligible patients will be randomized to the intensive structured monitoring (ISM) group or the active control (AC) group, with a glycosylated hemoglobin (HbA1c) target of <7.0%. Intervention will comprise (1) structured SMBG (4-point daily glucose profiles on 3 days per week [ISM]; discretionary, unstructured SMBG [AC]); (2) comprehensive patient education (both groups); and (3) clinician’s adjustment of diabetes medications using an algorithm targeting SMBG levels, HbA1c and hypoglycemia (ISM) or HbA1c and hypoglycemia (AC). The intervention and trial design build upon previous research by emphasizing appropriate and collaborative use of SMBG by both patients and physicians. Utilization of per protocol and intent-to-treat analyses facilitates assessment of the intervention. Inclusion of multiple dependent variables allows us to assess the broader impact of the intervention, including changes in patient and physician attitudes and behaviors. ClinicalTrials.gov (NCT00643474).
Similar content being viewed by others
Introduction
Type 2 diabetes is a growing, worldwide epidemic with significant clinical, social, and financial costs associated with the devastating microvascular and macrovascular complications of poorly controlled diabetes. Large randomized, controlled clinical trials, such as the Diabetes Control and Complication Trial (DCCT) and the United Kingdom Prospective Diabetes Study (UKPDS), have demonstrated that tight glycemic control reduces the risk of microvascular diabetic complications in both type 1 diabetes (T1DM) [1, 2], and T2DM [3–5]. Furthermore, long-term follow-up of the DCCT and UKPDS cohorts has shown the so-called legacy effect (i.e., a long-lasting positive effect of tight glucose control on diabetes complication), even if glucose control tends to deteriorate [2, 4]. However, in patients with a high cardiovascular risk, an intensive management of diabetes targeting normal glycated hemoglobin (HbA1c) levels with the use of multiple diabetes medications is associated with higher mortality than is a standard approach targeting higher HbA1c levels [6, 7].
While elevated HbA1c levels are clearly linked with the development and progression of diabetic complications, a growing body of evidence suggests that excessive post-prandial glycemic excursions may also be a significant risk factor for both microvascular [8] and macrovascular disease [9–14]. In patients with impaired glucose tolerance or T2DM post-challenge glucose and glycemic spikes are more strongly associated with carotid intima-media thickening than fasting glucose and HbA1c [12, 14]. Conversely, diabetes medications that limit post-prandial glucose excursions have been shown to reduce the risk of macrovascular disease [15–17]. Furthermore, in poorly controlled, non-insulin-treated T2DM patients, Monnier and colleagues [18] found a correlation between the production of 8-iso prostaglandin F2 (8-iso PGF2a), a marker of oxidative stress, and the magnitude of glucose fluctuations, calculated as the mean amplitude of glucose excursion (MAGE) [19]. This finding is important, given the possible role of post-prandial hyperglycemia in the pathogenesis atherosclerosis in patients with diabetes.
Unlike HbA1c measurement, which only presents an assessment of mean glucose over a 2–3 month period, SMBG detects intra-day glycemic variability, thus, providing valuable feedback on the impact of behavioral and pharmacological interventions on blood glucose levels. This information can assist clinicians in their therapeutic decision making when adjustments are needed, and it can increase patients’ understanding of their disease, reinforce self-management practices, and enhance patient empowerment [20, 21].
SMBG is only useful, however, when the glucose data are collected in a structured manner, accurately interpreted, and utilized to take appropriate therapeutic actions. New guidelines from the International Diabetes Federation (IDF) recommend that structured SMBG be performed over short periods of time, initially and periodically, in order to obtain glucose profiles that are representative of daily glucose excursions [20]. The aforementioned studies regarding glycemic variability [12, 14–17] suggest that SMBG should be performed with increased frequency to monitor glycemic excursions in T2DM patients even if the effect on HbA1c may be minimal. They also suggest that the various therapeutic strategies now in use should be evaluated for their potential to minimize glucose excursion, as well as for their ability to reduce HbA1c.
Although the value and utility of SMBG has been demonstrated in T1DM [1, 22] and insulin-treated T2DM [23, 24], SMBG use in non-insulin-treated diabetes remains somewhat controversial. Some studies have shown little or no glycemic benefit associated when SMBG was unstructured and/or clinicians underutilized results [25–27]; however, inherent limitations in the design of these studies has resulted in some questioning of the related findings in terms of external validity [20, 28, 29]. In essence, these studies were not designed to make use of SMBG to guide patient’s self-management and medical care with the aim of improving metabolic control. Of note, more recent studies, which utilized structured glucose monitoring regimens as an integral component of comprehensive treatment interventions, have demonstrated that appropriate use of SMBG facilitates therapy optimization and desired behavioral changes, leading to improved clinical outcomes [30–34].
With the aim of further addressing this issue, we designed a 12-month, prospective, multicenter, open, parallel group, randomized, and controlled clinical trial to evaluate the impact of structured SMBG on glycemic control in non-insulin-treated T2DM patients. In this study, the SMBG was used to generate information on glucose levels at specific times during the day and to make use of this information in order to prompt appropriate therapeutic actions by patients and clinicians.
Methods
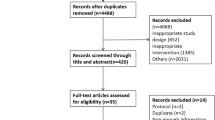
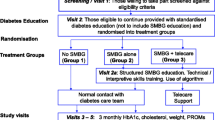
The Prospective, Randomized Trial on Intensive SMBG Management Added Value in Non-insulin-Treated T2DM Patients (PRISMA) is a 12-month, prospective, multicenter, open, parallel group, randomized, and controlled clinical trial that was designed to evaluate the added value of an intensive, structured SMBG regimen in T2DM patients treated with oral agents and/or diet alone (Fig. 1).

Diagram of the PRISMA study protocol
Patients randomized to the experimental group (intensive structured monitoring [ISM]) will be compared with patients who perform discretionary, unstructured SMBG (active control [AC]). Patients will be evaluated every 3 months with the same scales and measures over the 12-month study period. The study protocol, written in compliance with the Good Clinical Practice and the Helsinki Declaration [35], was first approved by the Ethics Committee of the San Raffaele Scientific Institute in Milano and then by the Ethics Committee of each participating clinical site. The clinical trial was registered at ClinicalTrials.gov (NCT00643474).
Study population
Participants will be recruited from 39 clinical sites in Italy, including diabetes outpatient clinics affiliated with academic institutions and hospitals of the Italian National Health Care System. Inclusion criteria are as follows: age between ≥ 35 and ≤75 years; non-insulin-treated T2DM (with diagnosis ranging from 1 to 10 years prior to study enrollment); treatment with diet and oral hypoglycemic agents or diet only; HbA1c ranging from 7.0% to 9.0% measured in the clinical site’s laboratory within 2 weeks before the date of signature of the informed consent; willingness to adhere to study procedures; ability and willingness to perform SMBG regimen; and willingness to sign the study informed consent. Exclusion criteria are as follows: T1DM, gestational diabetes or secondary diabetes; a history of insulin treatment for more than 7 consecutive days; previous intensive SMBG management (systematic or structured SMBG to adjust lifestyle or pharmacologic therapy); impending complications of diabetes; clinically significant, acute diseases (cardiovascular, gastrointestinal, neurological, genitourinary, and hematological systems) or severe uncontrolled hypertension (systolic blood pressure >180 mm Hg; diastolic blood pressure >100 mm Hg); serious diseases or limited life expectancy that would make intensive glycemic control inappropriate; known pregnancy or intention to become pregnant during the study; lactation; inability to read or to write; and potentially poor adherence to study procedures.
Intervention
The study intervention (Table 1) will last 12 months and will comprise three key components: (1) structured SMBG; (2) comprehensive patient education; and (3) clinician’s use of a diabetes medication algorithm. Combining these three components effectively addresses our study hypothesis.
Structured SMBG
A key component of the study intervention is utilization of a structured SMBG regimen that requires patients to generate 4-point daily glucose monitoring profiles on 3 days per week (2 working days, 1 weekend day). The measurements will be taken before breakfast, before lunch (pre-prandial), 2 h after lunch (post-prandial), and 5 h after lunch but before dinner (post-absorptive phase). All patients will complete one 3-day, 4-point glucose profile during the assessment period. ISM patients will be instructed to complete one 3-day, 4-point profile per week during the course of the study. SMBG data will be downloaded to a PC through a wireless device (Accu-Chek® Smart-Pix system, Roche Diagnostics, S.p.A., Monza, Italy) and analyzed using ad hoc software that provides easy-to-read summary statistics, which clinicians will use in conjunction with HbA1c and hypoglycemia to adjust diabetes medications (Fig. 2). AC patients will be instructed to complete one 3-day, 4-point profile prior to their 6- and 12-month visits to obtain data for comparison with the ISM group; in the AC group, SMBG data will not be downloaded from the glucometer, nor will be used to make adjustment of diabetes medications, which will be based exclusively on HbA1c and hypoglycemic events (either self-reported or documented).

Data analysis system. The print-out of the Smart-Pix device is organized in four boxes: Box 1 shows mean, standard deviation, and number of glucose measurements during the previous 4 weeks, by point of the daily profile presented as a bar graph; Box 2 shows mean and number of glucose measurements during the previous 4 weeks before breakfast and lunch and mean and number of post-prandial glycemic excursion, each presented as a speed dial: the pointer in the green zone indicates desirable values, in the yellow zone values that requires attention, and in the red zone values that require corrective action; the number of hypoglycemic episodes is reported in a dot that changes color from green (no glucose value <70 mg/dl) to red when blood glucose values <70 mg/dl are measured; Box 3 shows the low blood glucose index (LBGI) and high blood glucose index (HBGI) [40] and the average daily risk range (ADRR) [49] calculated on the glucose values since the last visit and presented as a bar graph in green color if in the desired range, in red if outside the desired range; and Box 4 shows the suggested changes in diabetes medication according to the algorithm presented in Fig. 3
Comprehensive patient education
A commercially available educational program (Accu-Chek® eduCare, Roche Diagnostics, S.p.A., Monza, Italy) will be used to provide standardized information to the patients who are enrolled in the study. The program is organized into subject-specific modules and includes charts and other materials to support training sessions and patient engagement. A basic session, covering nutrition, physical activity, SMBG, and diabetes medications, will be provided to all enrolled patients at the beginning of the study in order to avoid diabetes education differences between the ISM and AC group. Patients will complete additional modules of the educational program throughout the study. The educational program will be provided to study participants either by the investigator, the diabetes nurse, or the dietician.
Diabetes medication algorithm
During the study visits, clinicians will prescribe diabetes medication with the aim of reaching target HbA1c levels of <7.0% [36]. For ISM patients, clinicians will use an algorithm based on guidelines from international and national scientific societies (American Diabetes Association [ADA], European Association for the Study of Diabetes [EASD], International Diabetes Federation [IDF], Società Italiana di Diabetologia [SID], and Associazione Medici Diabetologi [AMD]) [37–39]. Incretin mimetics and DPP-4 inhibitors were not available in Italy when the PRISMA protocol was written and, therefore, were not included in the diabetes medication algorithm. Over the study period, exenatide, liraglutide, sitagliptin, vildagliptin, and saxagliptin became available for the treatment of patients with T2DM and investigators were notified that those drugs could be used in PRISMA participants according to the European Medicines Agency (EMA) therapeutic indications.
The algorithm bases the changes in diabetes medications (either type or dosage) on the mean fasting or pre-prandial glucose, on the difference between post- and pre-prandial glucose, and on hypoglycemic events (self-reported or documented) (Fig. 3). However, clinicians will have the option to make changes to the type or dosage of diabetes medications according to their own clinical judgment. Furthermore, clinicians will have the opportunity to use a feature of the ad hoc software that suggests the appropriate changes in diabetes medication according to the algorithm (Fig. 2). In the AC group, where patients are not required to perform any structured SMBG, the changes in diabetes medications will be based exclusively on HbA1c levels and hypoglycemic events (either self-reported or documented).

Diabetes medication algorithm. The diabetes medication algorithm is based on guidelines by international and national scientific societies [American Diabetes Association (ADA), European Association for the Study of Diabetes (EASD), International Diabetes Federation (IDF), Società Italiana di Diabetologia (SID) and Associazione Medici Diabetologi (AMD)] [37–39]
Adherence of care provider to the study protocol will be enhanced by training sessions before recruitment of patients and throughout the study. Three investigator’s meetings will be conducted during the study, and periodic newsletters will be e-mailed to investigators encouraging them to share knowledge and discuss study procedures.
Primary and secondary endpoints
Two primary endpoints will be tested subsequently, according to the following hierarchical order: (1) the change in HbA1c levels from baseline to visit 5; and (2) the percentage of participants reaching or maintaining the risk target (Low Blood Glucose Index (LBGI) ≤ 2.5 together with High Blood Glucose Index (HBGI) ≤ 5) from baseline to visit 5. HbA1c will be measured by a central laboratory (Laboraf Diagnostica e Ricerca, Milano, Italy) using the Variant II testing systems (Bio-Rad, Segrate, Italy) on whole blood samples kept frozen at −80° C until assayed. This method is certified by the National Glycohemoglobin Standardization Program as traceable to the method used in the DCCT.
LBGI and HBGI are computed using capillary glucose values according to Kovatchev et al. [40]. LBGI and HBGI have been shown to predict the risk of hypoglycemia and hyperglycemia, respectively [41–43].
The primary endpoints will be managed using a “sequence of comparisons” approach: step 1—shows that the ISM is superior to the AC group in the first co-primary endpoint; and step 2—shows that the ISM is superior to the AC in the second co-primary endpoint. Step 1 is based on a two-sided test with an alpha of 5%, and a significant result in the relevant direction is required to continue with step 2, which is also based on a two-sided test with an alpha of 5%.
Secondary endpoints in both study groups include changes in HBGI and LBGI, changes in SMBG frequency, changes in diabetes therapy (either type of medication or dosage), frequency and severity of hypoglycemic episodes, and changes in blood pressure, estimated glomerular filtration rate (eGFR, computed using a creatinine-based formula) [44] lipid profile, and body mass index (BMI). Additional secondary endpoints are the changes of the scores of two questionnaires: (1) a diabetes-specific quality of life questionnaire, which is a modification of the diabetes quality of life questionnaire used in the DCCT [45]. The original DCCT questionnaire was translated into Italian, modified for being used in patients with T2DM, and validated [46]; and (2) the diabetes-specific locus of control questionnaire [47] translated into Italian [48]. Study-related adverse events and diabetes-related adverse events will be recorded and compared in the two groups.
Additional secondary endpoints will be analyzed in the ISM group. These will include inter-visit changes in mean blood glucose (to assess glycemic exposure), blood glucose standard deviation (to assess glycemic variability), average fasting, pre-prandial, post-prandial, and post-absorptive phase glucose levels. The average difference between pre-prandial and post-prandial values within the same day and changes in lifestyle will also be analyzed. Correlations between patient age, diabetes duration, HbA1c, LBGI, HBGI, average daily risk range (ADRR) [49], mean blood glucose, standard deviation, and lipid levels will also be performed.
Changes in urinary 8-iso PGF2α, an index of oxidative stress, will be measured and analyzed for the study participants of the six clinical sites that agreed to collect 24-h urine samples at visit 1 and visit 5 [24]. One hundred microliters of a 0.005% butylhydroxytoluene solution in ethanol will be added to 10 milliliter of urine for each 24-h urine collection. Samples will be stored at −80° C until shipment in dry ice to the central laboratory. Urinary 8-iso PGF2α will be measured using an enzymatic immunoassay (Cayman Chemical Company, Ann Arbor, MI, USA) [50].
Sample size
Five hundred participants in each group (1,000 total) will be required to have an approximate 90% probability of detecting as significant (at the two-sided 5% level) a 0.3% difference between the ISM and AC group in the mean HbA1c change at visit 5 compared to baseline, with an assumed standard deviation of 1.25% and a loss to follow-up of 25%. This sample size will also provide 85% power to show an 11% difference between the two groups in percentage of subjects reaching or maintaining the risk target (LBGI ≤2.5 and HBGI ≤5), considering 50% of subjects arriving to target in the standard group.
Study procedures
The duration of the intervention is 12 months, with patient visits occurring during the assessment period at baseline (visit 1) and at months 3, 6, 9, and 12 (visit 2, 3, 4, and 5, respectively). The study will be managed by a certified clinical research organization (CROM, Verona, Italy).
Assessment period
During the assessment period—less than 15 days prior to the baseline visit—clinicians will describe the study in details to prospective participants, obtain written informed consent, record demographics, collect relevant medical history/lifestyle information, document all current medications, perform physical examinations, measure and record weight, height, blood pressure, heart rate, compute body mass index (BMI), and collect laboratory samples to measure lipid levels and serum creatinine. Urine for the measurement of 8-iso PGF2α will be collected in patients from the six clinical sites that agreed to participate in this ancillary study.
All eligible patients will be asked to complete the diabetes-specific quality of life questionnaire and the diabetes-specific locus of control questionnaire. They will receive a blood glucose meter (Accu-Chek® Aviva, Roche Diagnostics, Indianapolis, IN, USA) and 12 test strips to obtain a 4-point daily glucose profile 3 times in 1 week, two during working days and one during the weekend, and they will be scheduled to attend the basic session of the comprehensive educational program.
First visit
At the first visit, investigators will download SMBG data from the blood glucose meters using an automated downloading device. All serious and non-serious adverse events will be recorded. Eligible patients will then be randomized to the ISM or AC group. A computer generated randomization list prepared by a statistician and unknown to the investigators at the clinical sites will be used. The randomization code was developed by a statistician of the clinical research organization managing the study and will be unknown to the investigators at the clinical sites. A computer random number generator was used to select random permuted blocks of size 4. Randomization will be stratified by the diabetes treatment regimens of participants at the time of enrollment (diet only, or diet plus diabetes medications). Each clinical site will receive two sets of sealed, sequentially numbered, opaque envelopes containing the allocation information that were prepared by the personnel of the clinical research organization managing the trial. The investigators will be trained to write the name of the patient to be randomized on the envelope with the lowest sequence number of the appropriate set (diet only or diet plus diabetes medications) and then open the envelope and discuss treatment allocation with the patient. Investigators and study participants will not be blinded to group assignment. Statisticians analyzing the data and personnel at the central laboratory measuring HbA1c will be blinded to group assignment.
ISM patients will receive 150 test strips to obtain the requested glucose profiles and an additional 50 test strips for discretionary use (in case of hypoglycemia and/or at any time, they feel the need to monitor their blood glucose levels). They will also receive additional instructions on how to interpret SMBG data and a dedicated diary listing the assigned glycemic targets: <110 mg/dL for fasting and before-lunch values and <50 mg/dL difference between post-prandial and pre-prandial glucose levels) and suggestions on how to reach them. Suggestions will be in the form of “cheat lists” with appropriate lifestyle changes in response to real life situations (e.g., too high post-prandial glycemic excursion or a low pre-prandial glucose value, etc.).
AC patients will receive 24 test strips to obtain the requested glucose profiles and an additional 26 test strips for discretionary use. No instruction will be provided to AC patients regarding timing and frequency of testing.
Blood samples for the measurement of HbA1c by the central laboratory will be collected, frozen at −80°C, and stored until shipment in dry ice. All participants will be made aware of their risk of hypoglycemia, based on the diabetes medications they have been prescribed. After resolving any hypoglycemic event, patients will be asked to record the glucose value in an ad hoc diary and contact the center of case of additional hypoglycemic episodes close in time.
Follow-up visits
At each follow-up visit (months 3, 6, 9, and 12), investigators will perform physical examinations; measure and record BMI, blood pressure, and heart rate; collect blood samples for HbA1c measurements; and record changes in diabetes treatment. With patients randomized to the ISM group, investigators will review and discuss the patient’s diary, record changes in diet and physical activity, and review structured SMBG measurements. For patients in the ISM group, investigators will have the option to use the defined algorithm for adjusting diabetes medications based on the downloaded SMBG data; however, they will be allowed to make adjustments according to their clinical judgment. In the AC group, SMBG data will not be used to make adjustments of diabetes medications, which will be based exclusively on HbA1c and hypoglycemic events (either self-reported or documented).
ISM patients will receive 200 test strips at each follow-up visit for the requested glucose profiles (150 strips) and additional testing (50 strips). AC patients will receive “memo cards” at month 3 and 9 visits, reminding them to complete their 3-day, 4-point glucose profiles prior to month 6 and 12 visits, when data collected in the meters will be downloaded. All patients will be offered additional education sessions from the standardized education program throughout the study.
Statistical analysis
Primary and secondary endpoints will be analyzed for the intent-to-treat (ITT) population, which consists of all randomized patients who have a baseline and at least one post-baseline assessment. Only the primary endpoint will be analyzed for the per protocol (PP) population, which consists of all randomized patients from the ITT population who complete the study without major protocol violations and who are compliant with the SMBG regimen (i.e., for participant in the ISM group at least 80% of the expected structured capillary glucose measurements; for the AC group <200 unstructured discretionary capillary glucose measurements).
All statistical analyses and data processing will be performed using SAS® Software (version 9.02, TS level 02M0) on a Windows 2000/XP Pro operating system. Descriptive statistics will be provided in summary tables by group according to the type of variable summarized. Standard quantitative statistics (N, mean, standard deviation, median, minimum, and maximum) will be presented for quantitative criteria. Frequency distribution (number of non-missing observations [N] and percentages [%]) will be presented for qualitative criteria.
The analysis of the first co-primary endpoint (i.e., the mean change in HbA1c levels from baseline to visit 5) will be analyzed using the SAS PROC MIXED procedure and will be based on a mixed linear model with randomized group, center, visit, and the randomized-group-by-visit interaction as fixed effects and baseline as covariate. An unstructured variance–covariance matrix will be used to model the correlation within each patient and between the four repeated measurements (i.e., over the post-baseline visits). Maximum likelihood estimates and 95% confidence intervals of the mean difference between randomized groups at 12 months will be calculated using the Newton–Raphson algorithm implemented in the SAS® Proc Mixed Procedure. A two-sided test with a P value ≤0.05 will be considered statistically significant.
The interaction between randomized group and center will be assessed. A two-sided p value ≤0.10 will be considered statistically significant for the test of interaction between randomized group and center. If a statistically significant interaction is observed, efforts will be made to determine whether and how the interaction may affect the comparisons between randomized groups. Taking into account that maximum likelihood estimates have less bias than single imputation methods (e.g., LOCF) for missing informative and are, by definition, unbiased under a missing completely at random (MCAR) and a missing at random (MAR) mechanism of “missingness,” the mixed linear model described above represents an ideal choice for handling missing data and correcting for the bias potentially caused by drop-out participants in this study [51].
The analysis of the second co-primary endpoint (i.e., the percentage of subjects reaching or maintaining the risk target at visit 5 compared to baseline) will be analyzed using the SAS PROC FREQ procedure and will be based on the Cochran–Mantel–Haenszel test controlling for clinical site effects.
Secondary endpoints will be analyzed according to the type of variable. Summary statistics and 95% confidence intervals will be calculated for mean changes (continuous variables) and for risk differences (categorical variables).
Discussion
Diabetes is a growing, worldwide epidemic with significant associated clinical, social, and economic costs. It is critical that healthcare resources committed to diabetes management are applied both effectively and efficiently.
The benefits of SMBG have been demonstrated in type 1 diabetes (T1DM) and insulin-treated type 2 diabetes (T2DM) [1, 2, 23]; however, findings from studies in non-insulin-treated T2DM have been inconsistent. In studies that utilized random or low-intensity blood glucose monitoring regimens and/or where the data generated were not used to make treatment decisions, SMBG showed little or no benefit [25–27]. More recent studies, however, have shown a significant benefit when SMBG is structured in a manner that yields easily discernable blood glucose patterns, and when those data are used to make appropriate therapeutic changes in lifestyle or diabetes medications [30–34].
Our study was designed to test the optimal use of SMBG to improve HbA1c and minimize glycemic excursions. With the inclusion criteria of an HbA1c from 7 to 9% we expect to capture participants who are representative of the Italian patient population attending a diabetes clinic and not meeting the target of good glycemic control [52]. It is important to note that our intervention emphasizes appropriate utilization of SMBG data by both patients and clinicians, focusing on the value of SMBG as a tool that enhances patients’ understanding of their disease, supports the modification of patient behavior toward a healthier lifestyle, and guides and supports therapeutic changes by both clinicians and patients. Utilization of PP analyses in conjunction with ITT will enable us to both identify obstacles to adherence and more fully assess the efficacy of the intervention. Inclusion of multiple dependent variables will allow us to assess the broader impact of the intervention, including changes in patient and physician attitudes and behaviors. Patient recruitment for the PRISMA study started in April 2008 and was completed in May 2010; results will be available in 2012.
References
Diabetes Control and Complications Trial Research Group (1993) The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 329:977–986
Nathan DM, Cleary PA, Backlund JY et al (2005) Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med 353:2643–2653
U.K. Prospective Diabetes Study Group (1998) Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 352:837–853
Holman RR, Paul SK, Bethel MA et al (2008) 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 359:1577–1589
Gaede P, Vedel P, Parving HH, Pedersen O (1999) Intensified multifactorial intervention in patients with type 2 diabetes mellitus and microalbuminuria: the Steno type 2 randomised study. Lancet 353:617–622
Gerstein HC, Miller ME, Byington RP et al (2008) Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 358:2545–2559
Riddle MC, Ambrosius WT, Brillon DJ et al (2010) Epidemiologic relationships between A1C and all-cause mortality during a median 3.4-year follow-up of glycemic treatment in the ACCORD trial. Diabetes Care 33:983–990
Shiraiwa T, Kaneto H, Miyatsuka T et al (2005) Post-prandial hyperglycemia is an important predictor of the incidence of diabetic microangiopathy in Japanese type 2 diabetic patients. Biochem Biophys Res Commun 336:339–345
Sorkin JD, Muller DC, Fleg JL, Andres R (2005) The relation of fasting and 2-h postchallenge plasma glucose concentrations to mortality: data from the Baltimore longitudinal study of aging with a critical review of the literature. Diabetes Care 28:2626–2632
Hanefeld M, Koehler C, Schaper F et al ((1999) Postprandial plasma glucose is an independent risk factor for increased carotid intima-media thickness in non-diabetic individuals. Atherosclerosis 144:229–235
Cavalot F, Petrelli A, Traversa M et al (2006) Postprandial blood glucose is a stronger predictor of cardiovascular events than fasting blood glucose in type 2 diabetes mellitus, particularly in women: lessons from the San Luigi Gonzaga Diabetes Study. J Clin Endocrinol Metab 91:813–819
Temelkova-Kurktschiev TS, Koehler C, Henkel E et al (2000) Postchallenge plasma glucose and glycemic spikes are more strongly associated with atherosclerosis than fasting glucose or HbA1c level. Diabetes Care 23:1830–1834
Esposito K, Nappo F, Marfella R et al (2002) Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans: role of oxidative stress. Circulation 106:2067–2072
Esposito K, Ciotola M, Carleo D et al (2008) Post-meal glucose peaks at home associate with carotid intima-media thickness in type 2 diabetes. J Clin Endocrinol Metab 93:1345–1350
Hanefeld M, Cagatay M, Petrowitsch T et al (2004) Acarbose reduces the risk for myocardial infarction in type 2 diabetic patients: meta-analysis of seven long-term studies. EurHeart J 25:10–16
Hanefeld M, Chiasson JL, Koehler C et al (2004) Acarbose slows progression of intima-media thickness of the carotid arteries in subjects with impaired glucose tolerance. Stroke 35:1073–1078
Esposito K, Giugliano D, Nappo F, Marfella R (2004) Regression of carotid atherosclerosis by control of postprandial hyperglycemia in type 2 diabetes mellitus. Circulation 110:214–219
Monnier L, Mas E, Ginet C et al (2006) Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. JAMA 295:1681–1687
Service FJ, Molnar GD, Rosevear JW et al (1970) Mean amplitude of glycemic excursions, a measure of diabetic instability. Diabetes 19:644–655
International Diabetes Federation Clinical Guidelines Taskforce and International SMBG Working Group (2009) Global guideline on self-monitoring of blood glucose in non-insulin treated type 2 diabetes. International Diabetes Federation. Accessed June 2010 at http://www.idf.org
Parkin CG, Davidson JA (2009) Value of self-monitoring blood glucose pattern analysis in improving diabetes outcomes. J Diabetes Sci Technol 3:500–508
Evans JM, Newton RW, Ruta DA et al (1999) Frequency of blood glucose monitoring in relation to glycaemic control: observational study with diabetes database. BMJ 319:83–86
Karter AJ, Ackerson LM, Darbinian JA et al (2001) Self-monitoring of blood glucose levels and glycemic control: the Northern California Kaiser Permanente Diabetes registry. Am J Med 111:1–9
Nathan DM, McKitrick C, Larkin M et al (1996) Glycemic control in diabetes mellitus: have changes in therapy made a difference? Am J Med 100:157–163
Farmer A, Wade A, Goyder E et al (2007) Impact of self monitoring of blood glucose in the management of patients with non-insulin treated diabetes: open parallel group randomised trial. BMJ 335:132
O’Kane MJ, Bunting B, Copeland M, Coates VE (2008) Efficacy of self monitoring of blood glucose in patients with newly diagnosed type 2 diabetes (ESMON study): randomised controlled trial. BMJ 336:1174–1177
Davidson MB, Castellanos M, Kain D, Duran P (2005) The effect of self monitoring of blood glucose concentrations on glycated hemoglobin levels in diabetic patients not taking insulin: a blinded, randomized trial. Am J Med 118:422–425
Polonsky W, Fisher L, Schikman C et al (2010) The value of episodic, intensive blood glucose monitoring in non-insulin treated persons with type 2 diabetes: design of the Structured Testing Program (STeP) study, a cluster-randomised, clinical trial [NCT00674986]. BMC Fam Pract 11:37. Accessed June 2011 at http://www.biomedcentral.com/1471-2296/11/37
Klonoff D, Bergenstal R, Blonde LSA et al (2008) Consensus report of the coalition for clinical research—self-monitoring of blood glucose. J Diabetes Sci Technol 2:1030–1053
Kempf K, Kruse J, Martin S, ROSSO-in-praxi (2010) A self-monitoring of blood glucose-structured 12-week lifestyle intervention significantly improves glucometabolic control of patients with type 2 diabetes mellitus. Diabetes Technol Ther 12:547–553
Bonomo K, De Salve A, Fiora E et al (2010) Evaluation of a simple policy for pre- and post-prandial blood glucose self-monitoring in people with type 2 diabetes not on insulin. Diabetes Res Clin Pract 87:246–251
Duran A, Martin P, Runkle I et al (2010) Benefits of self-monitoring blood glucose in the management of new-onset Type 2 diabetes mellitus: the St Carlos Study, a prospective randomized clinic-based interventional study with parallel groups. J Diabetes 2:203–211
Polonsky WH, Fisher L, Schikman CH (2011) Structured self-monitoring of blood glucose significantly reduces A1C levels in poorly controlled, noninsulin-treated Type 2 diabetes: results from the structured testing program study. Diabetes Care 34:262–267
Franciosi M, Lucisano G, Pellegrini F, Cantarello A, Consoli A, Cucco L, Ghidelli R, Sartore G, Sciangula L, Nicolucci A; for the ROSES Study Group (2011) ROSES: role of self-monitoring of blood glucose and intensive education in patients with Type 2 diabetes not receiving insulin. A pilot randomized clinical trial. Diabet Med 28(7):789–796, doi:10.1111/j.1464-5491.2011.03268.x
World Medical Association declaration of Helsinki (1997) Recommendations guiding physicians in biomedical research involving human subjects. JAMA 277:925–926
American Diabetes Association (2011) Standards of medical care in diabetes—2011. Diabetes Care 34(Suppl 1):S11–S61
Nathan DM, Buse JB, Davidson MB et al (2006) Management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy. Diabetes Care 29:1963–1972
International Diabetes Federation Clinical Guidelines Task Force (2005) Global guideline for type 2 diabetes. Int Diabetes Federat. Accessed June 2010 at http://www.idf.org
Diabetologia AMD-SId. Standard Italiani per la Cura del Diabete Mellito. 2010
Kovatchev BP, Straume M, Cox DJ, Farhy LS (2000) Risk analysis of blood glucose data: a quantitative approach to optimizing the control of insulin dependent diabetes. J Theor Med 3:1–10
Kovatchev BP, Cox DJ, Gonder-Frederick LA et al (1998) Assessment of risk for severe hypoglycemia among adults with IDDM: validation of the low blood glucose index. Diabetes Care 21:1870–1875
Kovatchev BP, Cox DJ, Gonder-Frederick L, Clarke WL (2002) Methods for quantifying self-monitoring blood glucose profiles exemplified by an examination of blood glucose patterns in patients with type 1 and type 2 diabetes. Diabetes Technol Ther 4:295–303
Kovatchev BP, Cox DJ, Kumar A et al (2003) Algorithmic evaluation of metabolic control and risk of severe hypoglycemia in type 1 and type 2 diabetes using self-monitoring blood glucose data. Diabetes Technol Ther 5:817–828
Levey AS, Bosch JP, Lewis JB, for the Modification of Diet in Renal Disease Study Group (1999) A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Ann Intern Med 130:461–470
Diabetes Control and Complications Trial Research Group (1988) Reliability and validity of a diabetes quality-of-life measure for the diabetes control and complications trial (DCCT). The DCCT research group. Diabetes Care 11:725–732
Trento M, Passera P, Borgo E et al (2004) A 5-year randomized controlled study of learning, problem solving ability, and quality of life modifications in people with type 2 diabetes managed by group care. Diabetes Care 27:670–675
Peyrot M, Rubin RR (1994) Structure and correlates of diabetes-specific locus of control. Diabetes Care 17:994–1001
Trento M, Tomelini M, Basile M et al (2008) The locus of control in patients with Type 1 and Type 2 diabetes managed by individual and group care. Diabet Med 25:86–90
Kovatchev BP, Otto E, Cox D et al (2006) Evaluation of a new measure of blood glucose variability in diabetes. Diabetes Care 29:2433–2438
Pradelles P, Grassi J, Maclouf J (1985) Enzyme immunoassays of eicosanoids using acetylcholine esterase as label: an alternative to radioimmunoassay. Anal Chem 57:1170–1173
Heyting A, Tolboom JT, Essers JG (1992) Statistical handling of drop-outs in longitudinal clinical trials. Stat Med 11:2043–2061
AMD Annals. Quality indicators of diabetes care in Italy. http://www.aemmedi.it. Accessed Nov 2011
Acknowledgments
The PRISMA Study is funded by Roche.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Scavini, M., Bosi, E., Ceriello, A. et al. Prospective, randomized trial on intensive SMBG management added value in non-insulin-treated T2DM patients (PRISMA): a study to determine the effect of a structured SMBG intervention. Acta Diabetol 50, 663–672 (2013). https://doi.org/10.1007/s00592-011-0357-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00592-011-0357-y