
Abstract
Aims/hypothesis
Metabolomics has provided new insight into diabetes risk assessment. In this study we characterised the human serum metabolic profiles of participants in the Singapore Chinese Health Study cohort to identify metabolic signatures associated with an increased risk of type 2 diabetes.
Methods
In this nested case–control study, baseline serum metabolite profiles were measured using LC-MS and GC-MS during a 6-year follow-up of 197 individuals with type 2 diabetes but without a history of cardiovascular disease or cancer before diabetes diagnosis, and 197 healthy controls matched by age, sex and date of blood collection.
Results
A total of 51 differential metabolites were identified between cases and controls. Of these, 35 were significantly associated with diabetes risk in the multivariate analysis after false discovery rate adjustment, such as increased branched-chain amino acids (leucine, isoleucine and valine), non-esterified fatty acids (palmitic acid, stearic acid, oleic acid and linoleic acid) and lysophosphatidylinositol (LPI) species (16:1, 18:1, 18:2, 20:3, 20:4 and 22:6). A combination of six metabolites including proline, glycerol, aminomalonic acid, LPI (16:1), 3-carboxy-4-methyl-5-propyl-2-furanpropionic acid and urea showed the potential to predict type 2 diabetes in at-risk individuals with high baseline HbA1c levels (≥6.5% [47.5 mmol/mol]) with an AUC of 0.935. Combined lysophosphatidylglycerol (LPG) (12:0) and LPI (16:1) also showed the potential to predict type 2 diabetes in individuals with normal baseline HbA1c levels (<6.5% [47.5 mmol/mol]; AUC = 0.781).
Conclusions/interpretation
Our findings show that branched-chain amino acids and NEFA are potent predictors of diabetes development in Chinese adults. Our results also indicate the potential of lysophospholipids for predicting diabetes.
Similar content being viewed by others

Introduction
Type 2 diabetes is a constellation of disorders precipitated by complex and poorly understood interactions between environmental and genetic factors leading to diminished insulin sensitivity and hyperinsulinaemia [1]. According to the newly released Diabetes Atlas from the International Diabetes Federation, a total of 415 million adults had diabetes globally in 2014, and this is expected to rise to 642 million by 2040 (www.diabetesatlas.org). Type 2 diabetes is often present years before becoming clinically apparent, and complications such as nephropathy and retinopathy may already be present at the time of diagnosis. Thus, identifying high-risk individuals and elucidating the underlying mechanisms are crucial for the development of effective strategies to prevent type 2 diabetes. Conventional clinical and blood biomarkers, such as BMI, fasting blood glucose or HbA1c levels, are well-established predictors but remain imperfect and provide limited insight regarding underlying pathophysiology. Therefore, novel approaches are needed to advance the understanding of the mechanisms of diabetes development.
Metabolomics is a rapidly evolving technology and offers a new avenue for identifying novel biomarkers by assessing large numbers of metabolites that are substrates and products in metabolic pathways. Recent metabolomics studies have suggested that certain metabolites and metabolite classes may be associated with the risk of obesity, insulin resistance and type 2 diabetes [2–15]. The Framingham Offspring Study, a 12-year prospective cohort study, has shown that increases in certain branched-chain amino acids (BCAAs) (such as leucine, isoleucine and valine) and aromatic amino acids (AAAs) (such as tyrosine and phenylalanine) could predict the incidence of type 2 diabetes [6]. Similar results have also been demonstrated in other studies in both Western and Asian populations [3–5, 11, 12]. Other studies have noted that alterations in NEFA and long-chain fatty acids [2, 3, 5, 8, 9, 14], carbohydrate derivatives [2, 13] and tricarboxylic acid (TCA) cycle intermediate metabolites [9, 10] may also be associated with an increased risk of diabetes. These observations provide novel insights into the underlying pathophysiology of diabetes and highlight the possibility that alterations in metabolites may help to identify at-risk individuals prior to the onset of diabetes, in addition to standard clinical biomarkers.
However, most of the current metabolomics studies have used a targeted approach, directed at specific candidate metabolites [15]. This approach restricts the potential to discover novel biomarkers and hitherto unknown pathways in diabetes development. Furthermore, the studies have been conducted predominantly in Western populations [15]. Data from Asian populations are sparse and limited to cross-sectional studies [12, 16], which are subject to reverse causation (metabolites may be altered because of hyperglycaemia), and preclude the inference of temporal and causal relationships. To our knowledge, there has only been one case–control study conducted in China [17]. The study was carried out in Chinese adults living in Shanghai with a limited sample size (n = 73 incident diabetes) [17].
Here we systematically investigated the relationships between serum metabolites and incident type 2 diabetes within the Singapore Chinese Health Study (SCHS) cohort using an integrated MS-based untargeted metabolomics approach.
Methods
Study population
The SCHS is a population-based prospective cohort that recruited 63,257 Chinese men and women aged 45–74 years in Singapore between 1993 and 1998. During the first follow-up visit (1999–2004), of the 52,325 respondents, 32,575 participants agreed to donate morning blood samples for research. The second follow-up visit was conducted between 2006 and 2010. Because of the logistical and funding constraints of this untargeted metabolomics study, we randomly selected 200 individuals who reported a diagnosis of diabetes at the second follow-up visit but who were free of diabetes, cardiovascular disease or cancer at blood collection (i.e. the first follow-up visit). Matched controls (n = 200) were randomly selected from participants who were free of diabetes, cardiovascular disease or cancer at both the first and second follow-up visits. The controls were matched (1:1) to cases for sex, age (±3 years) and date of blood collection (±6 months) because these factors are well-known to influence metabolic profiles [18, 19] and are thus potential confounders in the association between metabolites and diabetes risk. In Singapore, diagnostic testing for diabetes at the time of the follow-up visits was done according to the 1997 ADA criteria [20], which were based on symptoms plus random glucose concentrations, fasting glucose concentrations or 2 h glucose tolerance tests. We only included individuals with type 2 diabetes in this study. To reduce the possibility of undiagnosed diabetes, controls with HbA1c levels ≥6.0% (42.1 mmol/mol) at baseline were excluded and replacement controls were sought from the remaining eligible participants.
All participants voluntarily joined this study, provided written informed consent and completed questionnaires that provided demographic information including age, sex, lifestyle factors and medical history. The study protocol was approved by the institutional review boards at the National University of Singapore.
Serum collection and preparation
Antecubital venous blood samples (20 ml) were taken from consenting participants in the morning; fasting was not required before blood collection. After collection, the blood samples were immediately placed on ice for transportation and were processed within 6 h to obtain serum samples, which were subsequently stored at −80°C [21]. For metabolic profiling, archived serum samples were allowed to thaw at 4°C overnight. Thawed serum samples (100 μl) were diluted four times with ice cold methanol containing 30 μg/ml N-(9-fluorenylmethoxycarbonyl)-glycine as an internal standard. After centrifugation of the mixture at 20,817 g for 10 min at 4°C, the supernatant fraction was collected and divided into two parts: one (100 μl) for LC-MS analysis and one (10 μl) for GC-MS analysis after derivatisation with N-methyl-N-trimethyl-silyl-trifluoroacetamide. A pooled quality control (QC) sample was prepared by mixing equal amounts (10 μl) of each serum sample.
Metabolic profiling
Serum metabolic profiling analysis was performed as described previously [22] in the NUS Environmental Research Institute at the National University of Singapore. Briefly, LC-MS analysis was performed on an Agilent 1290 ultrahigh pressure LC system coupled to a 6540 quadrupole time-of-flight (Q-ToF) mass detector (Agilent, Santa Clara, CA, USA) equipped with an electrospray ionisation source, at both positive and negative ion modes. GC-MS analysis was performed on an Agilent 7683B Series Injector coupled to an Agilent 7890A Series GC system and a 7200 Q-ToF mass detector (Agilent). The stability of LC-MS and GC-MS analytical systems was evaluated using the pooled QC samples (ESM Fig. 1) [23], which indicated good reproducibility and stability during the whole procedure. Structural identification of metabolic candidates followed our protocol that has been described previously [22].
Data processing and analysis
The LC-MS and GC-MS data were exported as separate mzData files using Agilent MassHunter qualitative analysis software (version B.06; Agilent), and pretreated with the open-source software MZmine 2 (http://mzmine.github.io) for peak detection, peak alignment and peak area normalisation. After screening of the metabolic features using the ‘80% rule’ and replacement of the missing values (i.e. zeros) by 1/2 minimum [24, 25], a total of 4859 features were extracted from the LC-MS data (2574 from positive ion mode and 2285 from negative ion mode), and 2482 were extracted from the GC-MS data. The preprocessed LC-MS and GC-MS data were log-transformed and merged for subsequent analyses. During data extraction, we found that peak abundances in three mass spectra (one case and two controls) were significant lower than the others. After excluding these participants, 197 cases and 197 matched controls are included in this report. We compared cases vs matched controls using an orthogonal partial least squares discriminant analysis (OPLS-DA) and a paired t test for metabolic signature discovery. The false discovery rate (FDR) method was used to correct for multiple hypothesis testing and to reduce false positives [26]. Those metabolic features with variable importance in the projection (VIP) values >1.0 in the OPLS-DA model and p values <0.05 in the paired t test were considered to be significantly different between cases and controls. Subsequently, we performed a conditional logistic regression analysis to estimate the OR per SD increment and 95% CI for the association between each novel metabolite and type 2 diabetes risk, with adjustment for confounding factors. An exploratory analysis that used a composite metabolomic score was conducted. The metabolomic score was created by summing the tertile ranks of metabolites that were associated with type 2 diabetes [27]. We examined significant correlations using the Pearson correlation analysis. We conducted receiver operating characteristic (ROC) analyses to evaluate the predictive potential of identified metabolic signatures for diabetes, by adding the metabolites to a multivariable-adjusted logistic regression model with established diabetes risk factors as the basic model.
Results
Participant and blood sample characteristics
The 197 pairs of cases and controls were matched by age and sex. Compared with controls, cases had higher BMI at baseline, and were more likely to be current smokers and have a history of hypertension (Table 1). These three factors were included in subsequent analyses as potential confounders. No significant differences were found for other variables. At baseline, 93 cases had normal HbA1c values (<6.5% or <47.5 mmol/mol, ‘low-risk cases’) and the remaining 104 had high HbA1c levels (≥6.5% or ≥47.5 mmol/mol, ‘high-risk cases’). The HbA1c values in the controls were all <6.0% (42.1 mmol/mol).
Metabolic and pathway changes
The 197 cases and 197 controls were separated with little overlap in the OPLS-DA score scatter plot (Fig. 1a), indicating that serum metabolic profiles were different at baseline. A post hoc analysis stratified by baseline HbA1c, presented in Fig. 1b (low-risk cases and matched controls) and Fig. 1c (high-risk cases and matched controls), indicated that the separation between cases and controls was weaker for the low-risk cases. The VIP and t test analyses identified 51 metabolites that were significantly different between cases and matched controls (ESM Table 1). Specifically, BCAAs (leucine, isoleucine and valine), NEFA (palmitic acid, stearic acid, oleic acid and linoleic acid), carbohydrates (CDP-glucose, d-galactose and gluconate) and organic acids (acetoacetic acid, lactic acid, pyruvate and urea) were significantly higher at baseline in cases, whereas proline, serine and medium-chain acylcarnitines (C5, C8, C10 and C13) were significantly lower. In addition, an array of lysophospholipids were altered in cases compared with controls, including increased lysophosphatidylinositol (LPI) and lysophosphatidylglycerol (LPG), and decreased lysophosphatidylcholine (LPC) and lysophosphatidylethanolamine (LPE) species.

OPLS-DA score scatter plots show serum metabolic perturbations in cases compared with controls. (a) Cases (red dots, n = 197) compared with matched controls (green dots, n = 197). R2Y (cumulative) = 0.61, Q2 (cumulative) = 0.38, p = 1.09 × 10−15. (b) Cases with HbA1c <6.5% (red dots, n = 93) compared with matched controls (green dots, n = 93), R2Y (cumulative) = 0.47, Q2 (cumulative) = 0.13, p = 2.37 × 10−8. (c) Cases with HbA1c ≥6.5% (red dots, n = 104) compared with matched controls (green dots, n = 104), R2Y (cumulative) = 0.60, Q2 (cumulative) = 0.34, p = 5.8 × 10−16
Stratification by fasting status verified that fasting had no significant effect on metabolite levels (ESM Table 2). Furthermore, we found that fasting status had no significant impact on the separation of cases from controls (ESM Fig. 2). On the basis of these results and an online database of metabolic pathways (KEGG PATHWAY Database, www.genome.jp/kegg), an aberrant metabolic correlation network related to the risk of type 2 diabetes was established (Fig. 2).

Major differential serum metabolites and pathways between cases and controls. Glycerate-3P: glycerate 3-phosphate; Glycerone-P: glycerone phosphate. PC includes LPC species (17:0, 18:0, 18:2 and 20:5); PE includes LPE species (20:3 and 20:5); PG includes LPG (12:0); PI includes LPI species (16:1, 18:1, 18:2, 20:3, 20:4 and 22:6). †Not associated with risk of incident type 2 diabetes after adjustment
Associations between baseline metabolites and type 2 diabetes
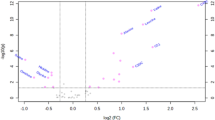
Of the 51 metabolites in the multivariate analysis, 35 were associated with an increased risk of diabetes at an FDR <0.05 (Table 2). The largest effect size was observed for glycine (OR 2.66 per SD increment [95% CI 1.72, 4.12]; p < 0.001). For the BCAAs, each SD increment in the log-transformed value was associated with a 44–66% increased odds of future diabetes (p = 0.001–0.015). The ORs for NEFA (palmitic acid, stearic acid, oleic acid and linoleic acid) and LPI species (16:1, 18:1, 18:2, 20:3, 20:4 and 22:6) were 1.43–2.10 per SD increment. In addition, the ORs across tertiles for each metabolite were generally consistent with the analyses using continuous variables (ESM Table 3). Specifically, the highest OR comparing extreme tertiles was for LPI (16:1) (4.45 [95% CI 2.10, 9.44]). The ORs comparing the extreme tertiles for BCAAs (leucine, isoleucine and valine) were 1.92–2.48 and NEFA (palmitic acid, stearic acid and oleic acid) and LPI species (18:1, 18:2, 20:4 and 22:6) were 2.06–3.82. The associations were generally consistent when stratified by fasting status with some differences in the point estimates (ESM Table 4). The metabolomic score created from the 35 metabolites ranged from 6 to 66, and the OR comparing extreme tertiles was 5.36 (95% CI 2.51, 11.43; p for trend <0.001; Fig. 3).

OR for type 2 diabetes by metabolomic score among cases (dark grey bars, n = 197) and controls (light grey bars, n = 197). Lines represent OR (95% CI) estimated using conditional logistic regression after adjustment for BMI, smoking status and history of hypertension. Solid line: OR (0.83–43.81); dotted lines: 95% CI low (0.47–6.26), high (1.46–306.48). Bars represent the percentage of participants within each group
Correlations between the 35 novel metabolites in the control group were assessed using the Pearson correlation analysis with adjustment for age, sex, BMI, smoking status and history of hypertension (ESM Fig. 3). Strong correlations were found within groups of related metabolites, such as BCAAs (mean correlation coefficient r = 0.777), NEFA (r = 0.703) and LPI species (r = 0.662). In addition, there was a strong correlation between BCAAs and NEFA (r = 0.638), but not with LPI species.
Predictive values of baseline metabolites
The diabetes cases were divided into low- and high-risk categories. The directions of the associations were generally consistent within the two subgroups, while some point estimates were different (ESM Table 5). Further stratification by fasting status among the 93 low-risk cases suggested again that fasting status had no substantial impact on the associations (ESM Table 6). To examine the performance of baseline metabolites for predicting type 2 diabetes, ROC curves were developed separately for the two groups, with adjustment for BMI, smoking status and history of hypertension as the basic model (Table 2). Six metabolites showed very good prediction capabilities (AUC > 0.80) in the high-risk case–control sets, including proline, glycerol, aminomalonic acid, LPI (16:1), 3-carboxy-4-methyl-5-propyl-2-furanpropionic acid (CMPF) and urea. Further, the c-statistic was increased from 0.769 in the basic model to 0.935 when a combination of the six metabolites was put into the model (Fig. 4a). It was found that LPG (12:0) and LPI (16:1) showed a fair potential for predicting type 2 diabetes in the low-risk case–control sets, with an AUC >0.75; a combined analysis increased the c-statistic from 0.728 to 0.781 (Fig. 4b).

ROC curves comparing the potential of different metabolites to predict type 2 diabetes. (a) Individuals at high risk (baseline HbA1c ≥6.5% [47.5 mmol/mol]. (b) Individuals at low risk (baseline HbA1c <6.5%)
Discussion
In this prospective study, we carried out a nested case–control study of diabetes within the SCHS cohort using an integrated LC-MS and GC-MS profiling analysis. We identified 35 metabolites that were associated with an increased risk of type 2 diabetes, including increased levels of BCAAs, NEFA and LPI species, and decreased levels of proline, glycerol and CMPF. Our results validated previous findings from a cross-sectional study, which demonstrated that diabetes patients had significantly raised amino acids (especially BCAAs such as leucine, isoleucine and valine) and NEFA (myristic acid, palmitic acid and stearic acid) when compared with controls [12]. Consistent with a recent study on the association of plasma metabolites with diabetes risk carried out in a Chinese population [17], we also found that baseline gluconate, pyruvate and valine levels were higher in cases compared with controls. Briefly, in the current study, the panel of 35 metabolic signatures can be broadly classified into three pathways: (1) carbohydrate metabolism; (2) amino acid metabolism; and (3) lipid metabolism.
Carbohydrate metabolism
In this study, we found that d-galactose was higher in cases than in controls and was strongly associated with type 2 diabetes. Galactose is a C-4 epimer of glucose and it can be rapidly converted to glucose through the Leloir pathway [28]. Increased galactose metabolism may lead to long-term, gradual increases in serum glucose and may contribute to insulin resistance. Serum glucose levels were associated with a higher risk of type 2 diabetes (OR comparing extreme tertiles 2.25 [95% CI 1.20, 4.21]; ESM Table 7). However, in the continuous variable analysis the association was not significant and the metabolite failed to meet the VIP or p value criteria, and thus glucose levels are not considered to be a novel clinical marker in the present study. As blood samples were not necessarily fasting, the association between glucose levels and type 2 diabetes may have been attenuated. The correlations between glucose level and the 35 novel metabolites are shown in ESM Table 8.
Amino acid metabolism
In 2011, the Framingham Offspring Study reported that elevated BCAAs (isoleucine, leucine and valine) and AAAs (tyrosine and phenylalanine) were able to predict an increased risk of diabetes up to 12 years prior to disease onset [6]. Several other studies have also identified BCAAs and AAAs as potent predictors of type 2 diabetes [3–5, 11, 12, 29, 30]. Consistent with these observations, our study found positive associations between three BCAAs and diabetes risk during the 6-year follow-up. The BCAAs are mainly derived from dietary intake and are metabolised in skeletal muscle and adipose tissue [31]. It has been reported that circulating amino acids may directly promote insulin resistance via disruption of insulin signalling in skeletal muscle [32]. Increased protein catabolism secondary to insulin resistance, coupled with impaired amino acid metabolism, contribute to elevated BCAAs [33].
Glycine is a major amino acid in mammals, and plays an important role in metabolic regulation, anti-oxidative reactions and neurological function [34]. Glycine is glucogenic, since one of its major metabolic routes involves conversion to serine; a large fraction of serine is converted into glucose, mainly through conversion to pyruvate by deamination catalysed by serine dehydratase, pyruvate is then converted into glucose by gluconeogenesis [35]. In this study, we found that glycine and pyruvate were significantly higher, and serine was significantly lower, in cases at baseline compared with controls. This suggests that increased activity in the glycine-serine-glucose pathway may account for diabetes risk. Animal models of diabetes support this theory, as the activity of serine dehydratase was increased in diabetic rats [36].
Lipid metabolism
Recent studies have identified fatty acids as independent predictors of progression to diabetes. Fatty acids impair insulin action via the Randle cycle, accumulation of intracellular lipid derivatives (e.g. diacylglycerol and ceramides), oxidative stress, inflammation and mitochondrial dysfunction [37]. We found palmitic, stearic, oleic and linoleic acids to be significantly elevated in cases compared with controls at baseline. This is consistent with the results of our previous cross-sectional study [12].
LPI belongs to the class of glycerophospholipids and is generated by phosphatidylinositol (PI) hydrolysis. LPI is involved in numerous physiological actions that are closely related to adipose tissue, including reproduction, angiogenesis, apoptosis and inflammation [38]. In addition, it is well accepted that LPI is an endogenous ligand of the G-protein-coupled receptor 55 (GPR55), which is an atypical cannabinoid receptor [39]. Recent studies have indicated that the LPI/GPR55 system is a novel target in obesity, and circulating plasma LPI levels are significantly increased in obese individuals [40]. To our knowledge, there are no available data on the association between the LPI/GPR55 system and the risk of type 2 diabetes. This is the first study to demonstrate significantly elevated levels of six LPI species (16:1, 18:1, 18:2, 20:3, 20:4 and 22:6) at baseline in individuals with diabetes compared with controls. Our findings indicate that the LPI/GPR55 system is particularly important in diabetes development, and further studies are required to investigate its potential role.
Carnitine transports activated long-chain fatty acids from the cytosol into mitochondria in the form of acylcarnitines and is therefore essential for fatty acid oxidation (energy supply). Carnitine also facilitates the removal of short- and medium-chain fatty acids that accumulate during normal metabolic processes from the mitochondria. Previous studies have demonstrated significantly increased free carnitine and acylcarnitine levels in patients with type 2 diabetes compared with healthy controls, which may be linked to insulin resistance [3, 8, 41, 42]. In this study, we observed that free carnitine was accumulated in cases; however, a medium-chain acylcarnitine (C10) was associated with a lower risk of type 2 diabetes. It has been postulated that a defect in beta-oxidation may be associated with insulin resistance [42]. Defects in long-chain acyl-CoA dehydrogenase may lead to accumulation of long-chain acylcarnitines and lower levels of medium- and short-chain acylcarnitines, but elevated long-chain acylcarnitines were not seen in our data.
Clinical implications
In addition to unravelling the potential mechanisms for diabetes development, our study also revealed six metabolic signatures for predicting diabetes, including proline, glycerol, aminomalonic acid, LPI (16:1), CMPF and urea. When combined, these six metabolites show a powerful ability to discriminate between high-risk individuals and controls. In addition, our results indicate that combined LPG (12:0) and LPI (16:1) might serve as a potential indicator for diabetes in low-risk individuals (HbA1c level <6.5% (47.5 mmol/mol). Overall, our findings indicate that LPI (16:1) could be a useful marker for the detection of type 2 diabetes in both low- and high-risk individuals.
Strengths and limitations
The strengths of this study include the use of a well-characterised Singapore Chinese cohort and two well-established MS techniques. This study is among the first to investigate the associations between serum metabolic signatures and the risk of type 2 diabetes in Asian populations using an untargeted metabolomics platform. A growing number of studies have used MS as a tool for biomarker discovery, but these studies have been largely performed using either LC-MS or GC-MS, here we applied both techniques with the aim of hitting more metabolite targets. However, some limitations of our study need to be acknowledged. First, we did not require participants to fast before blood samples were collected. Food consumption may influence some circulating metabolites. However, we did not find significant differences in the identified metabolites between fasted and non-fasted samples within the control group. Moreover, our study and a previous publication [3] showed that there were no considerable effect modifications of fasting status on metabolite–diabetes associations. Second, our findings were observed in a single cohort with a limited number of cases and controls. The candidate metabolites from this study should be validated in an independent and larger replication sample of Chinese adults. Finally, some of the participants already had HbA1c levels >6.5% (47.5 mmol/mol) at the time of blood collection (1999–2004), which could be classified as diabetes according to contemporary criteria [43]. However, we did observe some differences in the associations between the metabolites and diabetes risk when we stratified the cases by baseline HbA1c status; therefore, future studies need to take the diabetes diagnostic criteria into consideration and evaluate the relationships between metabolites and different glycaemic variables.
Conclusions
In summary, we investigated the associations between metabolic signatures and the risk of type 2 diabetes using two highly sensitive and complementary MS-based untargeted metabolomics techniques. The present study has revealed several early metabolic signatures for type 2 diabetes, which might advance prediction and prevention of type 2 diabetes in Chinese populations. In addition to well-recognised BCAAs and NEFA previously reported in Western populations, we identified six LPI species that were significantly associated with diabetes risk. LPI (16:1) specifically emerges as a potential predictor for the development of diabetes. These findings were observed in a single, small cohort, prompting the need for independent validation in well-designed, large-scale studies in the future.
Abbreviations
- AAA:
-
Aromatic amino acid
- BCAA:
-
Branched-chain amino acid
- CMPF:
-
3-Carboxy-4-methyl-5-propyl-2-furanpropionic acid
- FDR:
-
False discovery rate
- GPR55:
-
G-protein-coupled receptor 55
- LPC:
-
Lysophosphatidylcholine
- LPE:
-
Lysophosphatidylethanolamine
- LPG:
-
Lysophosphatidylglycerol
- LPI:
-
Lysophosphatidylinositol
- OPLS-DA:
-
Orthogonal partial least squares discriminant analysis
- PI:
-
Phosphatidylinositol
- QC:
-
Quality control
- Q-ToF:
-
Quadrupole time-of-flight
- ROC:
-
Receiver operating characteristic
- SCHS:
-
Singapore Chinese Health Study
- TCA:
-
Tricarboxylic acid
- VIP:
-
Variable importance in the projection
References
Tabak AG, Jokela M, Akbaraly TN, Brunner EJ, Kivimaki M, Witte DR (2009) Trajectories of glycaemia, insulin sensitivity, and insulin secretion before diagnosis of type 2 diabetes: an analysis from the Whitehall II study. Lancet 373:2215–2221
Fiehn O, Garvey WT, Newman JW, Lok KH, Hoppel CL, Adams SH (2010) Plasma metabolomic profiles reflective of glucose homeostasis in non-diabetic and type 2 diabetic obese African-American women. PLoS One 5, e15234
Floegel A, Stefan N, Yu Z et al (2013) Identification of serum metabolites associated with risk of type 2 diabetes using a targeted metabolomic approach. Diabetes 62:639–648
Menni C, Fauman E, Erte I et al (2013) Biomarkers for type 2 diabetes and impaired fasting glucose using a nontargeted metabolomics approach. Diabetes 62:4270–4276
Suhre K, Meisinger C, Doring A et al (2010) Metabolic footprint of diabetes: a multiplatform metabolomics study in an epidemiological setting. PLoS One 5, e13953
Wang TJ, Larson MG, Vasan RS et al (2011) Metabolite profiles and the risk of developing diabetes. Nat Med 17:448–453
Zheng Y, Hu FB (2015) Comprehensive metabolomic profiling of type 2 diabetes. Clin Chem 61:453–455
Mihalik SJ, Goodpaster BH, Kelley DE et al (2010) Increased levels of plasma acylcarnitines in obesity and type 2 diabetes and identification of a marker of glucolipotoxicity. Obesity 18:1695–1700
Salek RM, Maguire ML, Bentley E et al (2007) A metabolomic comparison of urinary changes in type 2 diabetes in mouse, rat, and human. Physiol Genomics 29:99–108
Zhang X, Wang Y, Hao F et al (2009) Human serum metabonomic analysis reveals progression axes for glucose intolerance and insulin resistance statuses. J Proteome Res 8:5188–5195
Stancakova A, Civelek M, Saleem NK et al (2012) Hyperglycemia and a common variant of GCKR are associated with the levels of eight amino acids in 9,369 Finnish men. Diabetes 61:1895–1902
Xu F, Tavintharan S, Sum CF, Woon K, Lim SC, Ong CN (2013) Metabolic signature shift in type 2 diabetes mellitus revealed by mass spectrometry-based metabolomics. J Clin Endocrinol Metab 98:E1060–E1065
Drogan D, Dunn WB, Lin W et al (2015) Untargeted metabolic profiling identifies altered serum metabolites of type 2 diabetes mellitus in a prospective, nested case control study. Clin Chem 61:487–497
Liu L, Wang M, Yang X et al (2013) Fasting serum lipid and dehydroepiandrosterone sulfate as important metabolites for detecting isolated postchallenge diabetes: serum metabolomics via ultra-high-performance LC-MS. Clin Chem 59:1338–1348
Guasch-Ferre M, Hruby A, Toledo E et al (2016) Metabolomics in prediabetes and diabetes: a systematic review and meta-analysis. Diabetes Care 39:833–846
Tillin T, Hughes AD, Wang Q et al (2015) Diabetes risk and amino acid profiles: cross-sectional and prospective analyses of ethnicity, amino acids and diabetes in a South Asian and European cohort from the SABRE (Southall And Brent REvisited) study. Diabetologia 58:968–979
Yu D, Moore SC, Matthews CE et al (2016) Plasma metabolomic profiles in association with type 2 diabetes risk and prevalence in Chinese adults. Metabolomics 12:1–11
Yu Z, Zhai G, Singmann P et al (2012) Human serum metabolic profiles are age dependent. Aging Cell 11:960–967
Mittelstrass K, Ried JS, Yu Z et al (2011) Discovery of sexual dimorphisms in metabolic and genetic biomarkers. PLoS Genet 7, e1002215
American Diabetes Association (1997) Report of the expert committee on the diagnosis and classification of diabetes mellitus. Diabetes Care 20:1183–1197
Koh WP, Yuan JM, Sun CL et al (2003) Angiotensin I-converting enzyme (ACE) gene polymorphism and breast cancer risk among Chinese women in Singapore. Cancer Res 63:573–578
Lu YH, Huang C, Gao L et al (2015) Identification of serum biomarkers associated with hepatitis B virus-related hepatocellular carcinoma and liver cirrhosis using mass-spectrometry-based metabolomics. Metabolomics 11:1526–1538
Gika HG, Theodoridis GA, Wingate JE, Wilson ID (2007) Within-day reproducibility of an HPLC-MS-based method for metabonomic analysis: application to human urine. J Proteome Res 6:3291–3303
Bijlsma S, Bobeldijk I, Verheij ER et al (2006) Large-scale human metabolomics studies: a strategy for data (pre-)processing and validation. Anal Chem 78:567–574
Smilde AK, van der Werf MJ, Bijlsma S, van der Werff-van der Vat BJ, Jellema RH (2005) Fusion of mass spectrometry-based metabolomics data. Anal Chem 77:6729–6736
Benjamini Y, Yekutieli D (2001) The control of the false discovery rate in multiple testing under dependency. Ann Statist 29:1165–1188
Zheng Y, Yu B, Alexander D et al (2013) Associations between metabolomic compounds and incident heart failure among African Americans: the ARIC Study. Am J Epidemiol 178:534–542
Holden HM, Rayment I, Thoden JB (2003) Structure and function of enzymes of the Leloir pathway for galactose metabolism. J Biol Chem 278:43885–43888
Sas KM, Karnovsky A, Michailidis G, Pennathur S (2015) Metabolomics and diabetes: analytical and computational approaches. Diabetes 64:718–732
Renner S, Romisch-Margl W, Prehn C et al (2012) Changing metabolic signatures of amino acids and lipids during the prediabetic period in a pig model with impaired incretin function and reduced beta-cell mass. Diabetes 61:2166–2175
Layman DK (2003) The role of leucine in weight loss diets and glucose homeostasis. J Nutr 133:261s–267s
Krebs M, Krssak M, Bernroider E et al (2002) Mechanism of amino acid-induced skeletal muscle insulin resistance in humans. Diabetes 51:599–605
She PX, van Horn C, Reid T, Hutson SM, Cooney RN, Lynch CJ (2007) Obesity-related elevations in plasma leucine are associated with alterations in enzymes involved in branched-chain amino acid metabolism. Am J Physiol Endocrinol Metab 293:E1552–E1563
Wang W, Wu Z, Dai Z, Yang Y, Wang J, Wu G (2013) Glycine metabolism in animals and humans: implications for nutrition and health. Amino Acids 45:463–477
Hetenyi G Jr, Anderson PJ, Raman M, Ferrarotto C (1988) Gluconeogenesis from glycine and serine in fasted normal and diabetic rats. Biochem J 253:27–32
Rowsell EV, al-Tai AH, Carnie JA (1973) Increased liver l-serine-pyruvate aminotransferase activity under gluconeogenic conditions. Biochem J 134:349–351
Charles MA, Eschwege E, Thibult N et al (1997) The role of non-esterified fatty acids in the deterioration of glucose tolerance in Caucasian subjects: results of the Paris prospective study. Diabetologia 40:1101–1106
Choi JW, Lee CW, Chun J (2008) Biological roles of lysophospholipid receptors revealed by genetic null mice: an update. Biochim Biophys Acta 1781:531–539
Oka S, Nakajima K, Yamashita A, Kishimoto S, Sugiura T (2007) Identification of GPR55 as a lysophosphatidylinositol receptor. Biochem Biophys Res Commun 362:928–934
Moreno-Navarrete JM, Catalan V, Whyte L et al (2012) The l-α-lysophosphatidylinositol/GPR55 system and its potential role in human obesity. Diabetes 61:281–291
Adams SH, Hoppel CL, Lok KH et al (2009) Plasma acylcarnitine profiles suggest incomplete long-chain fatty acid beta-oxidation and altered tricarboxylic acid cycle activity in type 2 diabetic African-American women. J Nutr 139:1073–1081
Schooneman MG, Vaz FM, Houten SM, Soeters MR (2013) Acylcarnitines reflecting or inflicting insulin resistance? Diabetes 62:1–8
American Diabetes Association (2010) Diagnosis and classification of diabetes mellitus. Diabetes Care 33:S62–S69
Acknowledgements
We thank S.-H. Low, National University of Singapore, for supervising the SCHS fieldwork, and R. Wang, University of Pittsburgh Cancer Institute, for maintenance of the SCHS database. We also thank M.C. Yu, founding principal investigator of the SCHS. Some of the data were presented as an abstract at the 5th International Conference and Exhibition on Metabolomics meeting in Osaka, Japan in 2016.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
This study was supported by the National Medical Research Council, Singapore (NMRC/CIRG/1354/2013) and National Institutes of Health, USA (RO1 CA144034 and UM1 CA182876).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
CNO, WPK and AP conceived and designed the research. YL and YW conducted the metabolomics study and interpreted the data. TS, HWC and JMY assisted in statistical analysis. YL and YW wrote the manuscript. YL, CNO, TS, HWC, JMY, WPK and AP critically revised the manuscript for key intellectual content. All authors approved the final version of the manuscript. WPK and AP are responsible for the integrity of the work as a whole.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM
(PDF 884 kb)
Rights and permissions
About this article

Cite this article
Lu, Y., Wang, Y., Ong, CN. et al. Metabolic signatures and risk of type 2 diabetes in a Chinese population: an untargeted metabolomics study using both LC-MS and GC-MS. Diabetologia 59, 2349–2359 (2016). https://doi.org/10.1007/s00125-016-4069-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-4069-2