
Abstract
Aims/hypothesis
Glucose sensors often measure s.c. interstitial fluid (ISF) glucose rather than blood or plasma glucose. Putative differences between plasma and ISF glucose include a protracted delay during the recovery from hypoglycaemia and an increased gradient during hyperinsulinaemia. These have often been investigated using sensor systems that have delays due to signal smoothing, or require long equilibration times. The aim of the present study was to define these relationships during hypoglycaemia in a well-equilibrated system with no smoothing.
Methods
Hypoglycaemia was induced by i.v. insulin infusion (360 pmol·m−2·min−1) in ten non-diabetic subjects. Glucose was sequentially clamped at ∼5, 4.2 and 3.1 mmol/l and allowed to return to normoglycaemia. Subjects wore two s.c. glucose sensors (Medtronic MiniMed, Northridge, CA, USA) that had been inserted for more than 12 h. A two-compartment model was used to quantify the delay and gradient.
Results
The delay during the fall in plasma glucose was not different from the delay during recovery (8.3±0.67 vs 6.3±1.1 min; p=0.27) and no differences were observed in the ratio of sensor current to plasma glucose at basal insulin (2.7±0.25 nA·mmol−1·l) compared with any of the hyperinsulinaemic clamp phases (2.8±0.18, 2.7±0.021, 2.9±0.21; p=NS). The ratio was significantly elevated following recovery to normoglycaemia (3.1±0.2 nA·mmol−1·l; p<0.001).
Conclusions/interpretation
The elevated ratio suggests that the plasma to ISF glucose gradient was decreased following hypoglycaemia, possibly due to increased skin blood flow. Recovery from hypoglycaemia is not accompanied by a protracted delay and insulin does not increase the plasma to s.c. ISF glucose gradient.
Similar content being viewed by others


Introduction
Intensive insulin therapy with tight glucose control significantly reduces diabetic complications but increases the risk of hypoglycaemia [1]. Although the increased risk can be partially mitigated through the use of frequent finger-stick blood glucose measurements, these are inconvenient, and often not performed at optimal times. Continuous glucose monitoring offers an alternative for minimising hypoglycaemic risk, but such systems are commonly based on measurement of s.c. interstitial fluid (ISF) glucose rather than its concentration in blood or plasma. Clinical concerns regarding this approach include the lag time in equilibration of glucose across the capillary endothelial barrier [2], possible errors in sensor glucose resulting from changes in the plasma to ISF glucose gradient [3], and protracted recovery times following hypoglycaemia [4, 5]. Several groups have suggested that s.c. ISF glucose can fall in advance of plasma glucose if the fall in glucose is due to increased glucose uptake in peripheral tissues [6–8] or that the response time may be different for falling vs rising glucose signals [9].
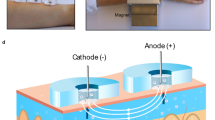
Assessment of ISF glucose kinetics is confounded by the lack of a direct method for its sampling. In the absence of such a measure, microdialysis has been the primary source from which ISF glucose has been determined [4, 6, 10, 11]. To this end, the glucose concentration in the effluent—i.e. the recovery—depends on the dialysis flow rate, the membrane pore size and the probe dimensions, making data interpretation difficult [6]. An alternative to microdialysis is to use an amperometric glucose sensor and analyse the data using a two-compartment model of plasma and ISF glucose [7, 8]. In the model (Fig. 1), ISF glucose is determined by the rate of transport of glucose across the capillary bed and the rate of glucose uptake into the surrounding cells. Sensor current is taken as proportional to ISF glucose. This approach was used in the present study to evaluate ISF glucose dynamics during insulin-induced hypoglycaemia.

Plasma ISF glucose equilibration model. Glucose (circles) is assumed to move from plasma to interstitial fluid in proportion to the concentration in each compartment (proportionality constants K 12 and K 21), to be subsequently cleared from the interstitial fluid by cellular glucose uptake. Cellular glucose uptake (K 02) may be increased by insulin (inverted triangles) binding to its receptor (notched square)
Subjects, materials and methods
Subjects
This study included ten non-diabetic subjects (six men, four women) aged 45±9 years (mean ± SD) with a body mass index of 27.6±5.6 kg/m2. Subjects were admitted to the University of California at Los Angeles (UCLA) General Clinic Research Center on the evening prior to experiments. Next morning, they underwent a three-step euglycaemic–hypoglycaemic–hyperinsulinaemic clamp. The protocol was approved by the UCLA Institutional Review Board, and all subjects gave written informed consent.
Protocol
Two Medtronic MiniMed (Northridge, CA, USA) s.c. glucose sensors were inserted in the abdominal area of each subject the evening before experiments (∼18.00 h). The Medtronic MiniMed s.c. sensor is an amperometric device utilising glucose oxidase and is used as a component of a continuous glucose monitoring system (CGMS; Medtronic MiniMed) [12]. On the morning following insertion, and after a 10- to 12-h overnight fast, an i.v. catheter was inserted in the antecubital vein of one arm for infusing insulin and glucose, and a second catheter was placed retrograde in a dorsal vein of the contralateral hand for blood withdrawal. The hand was placed in a heating pad to arterialise the blood and samples were collected at −30, −10 and 0 min and every 5 min thereafter until 380 min. At 0 min, insulin (360 pmol·m−2·min−1) and glucose (20%; variable rate) infusions were started. Plasma glucose was clamped at ∼5 mmol/l until 90 min, at ∼4.2 mmol/l until 180 min, and at 3.1 mmol/l until 270 min. Subsequently, insulin and glucose infusions were stopped, and plasma glucose was allowed to return to basal. In some subjects, the i.v. glucose infusion was continued to prevent plasma glucose from dropping below 3 mmol/l.
Biochemical measurements
Plasma glucose was measured with the glucose oxidase method using a Beckman glucose analyser (Beckman Instruments, Fullerton, CA, USA). Plasma insulin was measured by radioimmunoassay with reagents from Linco Research (St Louis, MO, USA).
Sensor calibration
To assess the impact of ISF glucose delay on real-time sensor performance, sensor glucose (S G) was calculated as:
where the calibration factor (CF; mmol·l−1·nA−1) was obtained from the current (I sig) and plasma glucose value at t=−30 min (one-point calibration). Sensors with CF greater than 1 mmol·l−1·nA−1 were taken to be non-responding and were excluded from all analysis (two sensors, in two individuals). This one-point calibration is, in principle, applicable for real-time sensor glucose calculations.
The mean absolute difference (MAD) between one-point calibrated sensor and plasma glucose (PG) was calculated as:
One-point calibration does not ensure that initial and final sensor glucose levels correspond exactly to the initial and final plasma glucose values during any individual excursion. For the delay to be estimated qualitatively, it is necessary that these values correspond. If the initial sensor glucose point is accurately obtained, overestimation of the final glucose value leads to underestimation of the delay and vice versa. Thus, to assess delay, two-point calibrations were performed separately for the fall and recovery in plasma glucose. For the two-point calibration, sensor glucose was calculated as:
where CF 2 is the incremental change in plasma glucose divided by the incremental change in sensor current and S 0 is the predicted sensor glucose at 0 current. For the fall in plasma glucose, CF 2 and S 0 were calculated from the time points −30 and 270 min, and for the recovery to normoglycaemia they were calculated from the time points 270 and 380 min.
Two-compartment sensor model analysis
While the two-point calibration provides a qualitative estimate of the ISF glucose delay, it does not allow the delay to be quantified. For this, the dynamic relationship between plasma glucose and sensor current was derived from the model shown in Fig. 1 (see Appendix for derivation of equations) and the delay was estimated using non-linear least squares (Mlab; Civilized Software, Bethesda, MD, USA). This analysis characterises delay as the time to reach 63% equilibration (50% equilibration can be calculated as T 1/2=0.693 times the delay and 95% equilibration as three times the delay). The analysis also provided a model-based estimate of sensor sensitivity (nA·mmol−l·l) that included the gradient in plasma to ISF glucose. Delay time and sensor sensitivity were estimated separately for both the fall (from −30 to 270 min) and rise (from 270 to 360 min) in plasma glucose.
Statistical analysis
Statistical analysis was performed using Prism version 3.0 (GraphPad Software, San Diego, CA, USA). Agreement between the one-point calibrated sensor signal and plasma glucose was evaluated by the paired t-test, linear regression, correlation and Clarke error grid [13]. For the regression analysis, 95% confidence intervals are reported for the estimates of slope and intercept. Analysis of variance was used to evaluate differences in the ratio of plasma glucose to I sig at each clamp period (basal, euglycaemic–hyperinsulinaemic, 4.2 mmol/l, 3.1 mmol/l, and after recovery). Post hoc analysis for differences among the clamp periods was performed using Dunnett’s test with the basal period as the control group. Power calculations were performed using Mlab. Data are reported as mean±SEM except where noted. Statistical significance and confidence intervals were calculated at the p<0.05 levels.
Results
On initiation of the insulin infusion, plasma insulin levels rapidly increased from basal (80.4±19.2 pmol/l) to near steady-state levels (736.2±68.4 pmol/l) within 15 min (Fig. 2a). Plasma glucose concentration was 5.8±0.14 mmol/l at basal, and was subsequently clamped at 5.5±0.06, 4.3±0.03 and 3.3±0.05 mmol/l (average of last half-hour for each clamp period). The peak glucose infusion rate (Fig. 2b) was 5.2±0.8 mg·kg−1·min−1 during euglycaemia–hyperinsulinaemia and was thereafter decreased. After discontinuing insulin and glucose infusions, plasma glucose and insulin concentrations returned to pre-clamp levels by 380 min (5.3±0.17 mmol/l and 70.2±12 pmol/l, respectively; not different from basal, p>0.05 for both).

a Plasma glucose (open circles; mean and SD) and insulin (closed squares; mean and SD) concentration, b glucose infusion rate and c sensor current (mean), during insulin-induced hypoglycaemia. Steady-state glucose was calculated during the last 30 min of each clamp phase (P1 to P5). Sensor current (solid line) was separately fitted for the fall (−30<t<270 min; fit shown by dotted line) and recovery (240<t<380 min; fit shown by dashed line) using the model of Fig. 1. Sensor current is shown with standard deviation bars removed for clarity
The plasma/ISF model (Fig. 1) described sensor current during both the fall to and recovery from hypoglycaemia (Fig. 2c; r 2=0.899±0.0177 and 0.928± 0.0197, respectively). However, there were brief periods when the sensor current was systematically under- or overestimated (residuals). For the fall, current was overestimated in the period −30<t<60 min and underestimated in the period 200<t<270 min; during recovery, it was overestimated in the period 240–320 min and underestimated in the period 320–380 min. These residuals were due to an increase in the model estimate of sensor sensitivity (2.9±0.2 vs 2.7±0.2 nA·mmol−1·l; p<0.05; rise vs fall, respectively). The increased sensitivity was also associated with a tendency for a smaller ISF glucose delay (6.3±1 vs 8.3±0.63 min; recovery vs fall) but the difference was not statistically significant (p=0.22). Post hoc analysis indicated that our data had 80% power to detect a difference in delay time of 3.8 min.
To assess changes in sensor sensitivity independent of any model analysis, the steady-state ratio of sensor current to plasma glucose was calculated during each of the clamp periods (Fig. 3). This ratio tended to increase during periods in which plasma glucose was clamped at ∼ 4.2 and 3.1 mmol/l and was significantly elevated following recovery to normoglycaemia (3.1±0.20 vs 2.7±0.25 nA·mmol−1·l; recovery period 5 vs basal period 1; p<0.05, ANOVA).

In vivo sensor sensitivity estimated from the steady-state ratio of sensor current to plasma glucose during the basal period (period 1; −30<t<0), the euglycaemia/hyperinsulinaemia period (period 2; 0<t<90 min), the first hypoglycaemic step (period 3; 90<t<180 min), the second hypoglycaemic step (period 4; 180<t<270) and the recovery period (period 5; 270<t<380). Steady state was defined for each period as the last half-hour. Following the clamp, sensor sensitivity was significantly higher than in the basal period (period 5 vs period 1; *p<0.05; ANOVA)
Using a single-point calibration, sensor glucose tracked the transient fall in plasma glucose during the euglycaemic–hyperinsulinaemic period (Fig. 4a; from 0 to 90 min). During the fall to ∼4.2 mmol/l (90 to 180 min), sensor glucose lagged plasma glucose but no difference was observed at steady state (from 150 to 180 min). For the fall to ∼3.1 mmol/l (from 180 to 270 min), sensor glucose again lagged plasma glucose and the steady state value was ∼0.2 mmol/l higher (3.5±0.11 vs 3.3±0.04; p=0.03). No lag in sensor response was observed during the return of plasma glucose to basal levels, but the final sensor glucose reading was ∼0.5 mmol/l higher than the plasma glucose level (5.9±0.010 vs 5.3±0.008 mmol/l; p<0.01). The lack of a perceived lag in sensor response during this phase was probably due to an increase in the apparent sensor sensitivity (i.e. the increase in current to glucose ratio). Thus, when two-point calibrations were separately applied to the fall and recovery, sensor glucose had similar lags during both the fall and recovery (Fig. 4b).

a Plasma glucose (open circles) together with sensor glucose (solid line) calculated using a one-point calibration at −30 min. Following a 30-min basal period (−30<t<0), plasma glucose was clamped at basal (0<t<90 min), 4.2 mmol/l (90<t<180 min) and 3.1 mmol/l (180<t<270 min) and then allowed to return to normal levels (270<t<380 min). The glucose scale is expanded to highlight differences in sensor vs plasma glucose. b Plasma glucose (open circles) and sensor glucose with separate two-point calibrations for the fall (short dashes) and rise (long dashes) in plasma glucose
The one-point calibration resulted in sensor and plasma glucose levels that were well correlated (Fig. 5a; r=0.87) with the regression slope (1.0±0.016, CI 0.99 to 1.1) and intercept (2.1±1.3, CI −0.51 to 4.7 mmol/l), not different from 1 and 0, respectively (p>0.05, both). Separately regressing the data during the fall and recovery resulted in slopes that were significantly different (0.94±0.016, CI 0.91 to 0.97, and 1.2±0.039, CI 1.1 to 1.3, respectively) with a positive intercept for the fall (7.8±1.3, CI 5.3 to 10 mmol/l) and negative intercept for the recovery (−9.5±3.3, CI −16 to −3.0 mmol/l; rising and falling intercepts different, p<0.05). Correlation coefficients were similar during the fall and recovery of plasma glucose (0.80 and 0.73, respectively). Clarke error grid analysis (Fig. 5b) indicated that 97.1% of the sensor glucose values were within the clinically acceptable A and B zones, with the remaining 2.9% in the clinically unacceptable D zone (no data were in zone C or E). The mean absolute difference between sensor glucose and plasma glucose levels averaged 10.7%, the mean sensor glucose being 0.2 mmol/l higher (p<0.05).

a Correlation of plasma glucose and sensor glucose during falling glucose (open inverted triangle) and rising (solid triangle) excursion. Sensor glucose was calculated using a one-point calibration (Fig. 4). Slope (1.0±0.016) and intercept (2.1±1.3 mmol/l) were not different from 1 and 0, respectively, when all data were included (solid line). Separate correlations for the fall (dotted line) and rise (dashed line) in plasma glucose indicated deviations from unity slope and zero intercept. b Clarke error grid. Zones A–D categorise errors based on clinical impact (A, accurate or no impact; B, acceptable treatment with benign impact; C, unacceptable treatment; D, failure to detect necessary treatment)
Discussion
This study has three main results. First, our data show that the plasma to s.c. ISF glucose gradient is not increased during insulin-induced hypoglycaemia. Second, changes in s.c. ISF glucose lag those in plasma glucose by 6–8 min irrespective of whether the glucose is increasing or decreasing. Third, differences between ISF and plasma glucose levels do not result in significant errors when the s.c. tissue is used as a site for glucose sensing.
The absence of changes in the sensor current to plasma glucose ratio during hyperinsulinaemia shows that insulin did not increase the plasma to ISF glucose gradient. This finding is in agreement with those from our previous hyperglycaemic clamp studies, which also showed no effect of insulin on the plasma to s.c. ISF glucose gradient or the delay time [7, 8]. Taken together, these results imply that s.c. ISF fluid is not derived from tissues that appreciably increase glucose uptake in response to insulin (i.e. not derived from the plasma of blood flowing to these tissue beds). It is more likely that s.c. ISF is derived from blood flowing to skin or s.c. fat. Skin is unlikely to increase glucose uptake in response to insulin, and the primary effect of insulin on s.c. fat is to suppress lipolysis [14]. Insulin levels used in the present study are unlikely to have resulted in more than 1–2% of the increased glucose disposal being taken up by the s.c. fat [15]. In the absence of an appreciable increase in glucose uptake into s.c. fat, it is unlikely that glucose levels in the ISF bathing this tissue would fall in advance of plasma glucose.
Several groups have postulated that ISF glucose could fall in advance of plasma glucose following insulin treatment [11, 16, 17]. This hypothesis, commonly referred to as ‘push–pull’, maintains that an increase in plasma glucose pushes glucose into the ISF space, and an increase in tissue glucose uptake lowers the ISF-glucose and pulls glucose from the plasma space [16, 17]. The hypothesis is consistent with the model of Fig. 1, provided the fall in plasma glucose is due to increased glucose uptake by the tissues bathed by the ISF fluid [2] (e.g. muscle). While our steady-state data do not support the ‘pull’ component of this hypothesis, residual errors in the ability of the ISF model to describe sensor current in the first 90 min of the insulin infusion were observed (Fig. 4). During this transition period, sensor glucose was lower than plasma glucose, which is consistent with glucose being pulled from the plasma space. Thus, the push–pull hypothesis cannot be completely dismissed.
Our results differ from studies showing sensor glucose lower than plasma glucose during hypoglycaemia [18], and from those showing a protracted delay during the recovery from hypoglycaemia [5, 19]. These discrepancies can probably be attributed to differences in the length of time sensors were allowed to stabilise, and to differences in data smoothing. In the present study, sensors were inserted ∼12 h before the clamp procedure. Studies in which sensors were inserted with less equilibration time [18, 19] may have interpreted the slow loss in sensor sensitivity typically observed for several hours following insertion as an increase in the plasma to ISF glucose gradient. Thus, in a hypoglycaemic clamp study in which sensors were inserted ∼16 h before the clamp, no steady-state difference in blood and sensor glucose was observed during hypoglycaemia [5]; the recovery was, however, delayed.
The studies reporting a protracted delay during recovery from hypoglycaemia [5, 18, 19] have all used the Medtronic MiniMed CGMS. However, this system applies data smoothing routines that limit the maximal rate of change in the sensor signal to that observed during physiological day-to-day glucose excursions. Unphysiological changes, such as those that can be obtained with i.v. glucose administration, are smoothed. The smoothing removes noise from the sensor signal, but invariably adds a delay to the response that is unrelated to ISF glucose equilibration. Although i.v. glucose infusions were also used in this study, the smoothing routines were removed. The absence of these smoothing routines in the present study could explain both the faster response times observed here and the absence of any substantial difference in delay during the fall vs rise in glucose (power calculations indicated a difference of 3.8 min would have been detectable). Importantly, if the delay is assessed during physiological changes in blood glucose (e.g. meals), the smoothing has very little effect, as was demonstrated in a study in which the delay was assessed during meals consumed by individuals with type 1 diabetes mellitus [20]. In the meal study [20], the delay (4–10 min) was similar to that reported here (6–8 min) and no difference was observed for rising and falling glucose concentrations.
Also in marked contrast to our findings are microdialysis studies showing an increase in the plasma to ISF glucose gradient during insulin-induced hypoglycaemia, and a much longer response time (up to 18 min [4]). However, microdialysis systems generally have intrinsic delays related to glucose equilibration across the microdialysis membrane itself. Full equilibration across the membrane often requires a slow dialysis perfusion rate (0.2–0.3 μl/min), which results in a long transit time (10–15 min), and the requirement that fractions be collected over finite time periods (15–60 min). An increase in plasma to ISF glucose gradient was observed with microdialysis during insulin-induced hypoglycaemia [4]; however, the fraction period was relatively short there (15 min). When longer fraction periods (30 min) were used in the same system, the gradient did not show an increase and the delay during a fall and rise in glucose concentration was thought to be similar [10].
In the present study, the ratio of sensor current to plasma glucose increased following the recovery from hypoglycaemia (Fig. 3). This implies either that the sensor’s sensitivity increased or that the gradient between plasma and s.c. ISF glucose decreased. It is unlikely that the sensor’s intrinsic sensitivity increased during the clamp as sensors typically lose sensitivity with time, although this possibility cannot be ruled out. It is possible, however, that the gradient between plasma and ISF glucose decreased during the period of hypoglycaemia. A decrease in the gradient can occur if cutaneous/subcutaneous blood flow increased or new capillaries were recruited. Cutaneous blood flow has been shown to increase during insulin-induced hypoglycaemia [21], and hyperinsulinaemia per se has been shown to cause capillary recruitment [22, 23]. These effects on blood flow may be reduced or absent in individuals with type 1 diabetes, but whether this could influence sensor performance is unknown.
While our data indicate that the ISF glucose delay was no more than 6–8 min, such delays still contribute to the glucose sensing errors. One-point calibration resulted in an MAD of ∼10% relative to plasma glucose (Beckman). Of these errors, ∼3% were in zone D of the Clarke error grid (Fig. 5b), which is deemed clinically unacceptable for a single self-monitored blood glucose reading. Errors in zone D were a direct consequence of the delay; that is, the values in zone D occurred during the period that plasma glucose had fallen below 3.9 mmol/l but the sensor glucose was lagging by 6–8 min (falling but still higher than 3.9 mmol/l). This lag also contributed to the bias in the regression equation (slope<1, intercept >0) observed during the fall (Fig. 5a). While the delay would also have contributed to the error during the rise, the bias in slope and intercept (slope >1, intercept<0) was more heavily influenced by the steady-state overestimation of sensor glucose following recovery to euglycaemia. Generally, errors resulting from delay can be minimised by signal processing algorithms [2, 24] but those due to steady-state differences in the plasma to ISF glucose gradient can only be corrected by recalibration. Nonetheless, in the absence of delay correction or recalibration, 97% of the sensor readings remained in the clinically acceptable Clarke A and B regions during the hypoglycaemic excursion.
In conclusion, the present study demonstrated that the ISF glucose-sensing site is not affected by changes in peripheral glucose uptake, and that the delay in s.c. ISF glucose equilibration during a fall to hypoglycaemia, or the subsequent recovery, is no more than 6–8 min. Delays of this magnitude are unlikely to limit the ISF site from being fully developed as an optimal site for estimating blood glucose changes and providing corrective measures, either through early patient warning or automation of insulin delivery.
Abbreviations
- CGMS:
-
continuous glucose monitoring system
- ISF:
-
interstitial fluid
- MAD:
-
mean absolute difference
- UCLA:
-
University of California at Los Angeles
References
DCCT (1993) The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. New Engl J Med 329:977–986
Rebrin K, Steil GM (2000) Can interstitial glucose assessment replace blood glucose measurements? Diabetes Technol Ther 2:461–472
Baron AD (1994) Hemodynamic actions of insulin. Am J Physiol 267:E187–E202
Moberg E, Hagström-Toft E, Arner P et al (1997) Protracted glucose fall in subcutaneous adipose tissue and skeletal muscle compared with blood during insulin-induced hypoglycaemia. Diabetologia 40:1320–1326
Cheyne EH, Cavan DA, Kerr D (2002) Performance of a continuous glucose monitoring system during controlled hypoglycaemia in healthy volunteers. Diabetes Technol Ther 4:607–613
Wientjes KJ, Schoonen AJ (2001) Determination of time delay between blood and interstitial adipose tissue glucose concentration change by microdialysis in healthy volunteers. Int J Artif Organs 24:884–889
Rebrin K, Steil GM, Van Antwerp WP et al (1999) Subcutaneous glucose predicts plasma glucose independent of insulin: implications for continuous monitoring. Am J Physiol 277:E561–E571
Steil GM, Rebrin K, Mastrototaro J et al (2003) Determination of plasma glucose during rapid glucose excursions with a subcutaneous glucose sensor. Diabetes Technol Ther 5:27–31
Kulcu E, Tamada JA, Reach G et al (2003) Physiological differences between interstitial glucose and blood glucose measured in human subjects. Diabetes Care 26:2405–2409
Moberg E, Hagström-Toft E, Bolinder J (1997) Detection of hypoglycaemia by microdialysis measurements of glucose in subcutaneous adipose tissue. Horm Metab Res 29:440–443
Sternberg F, Meyerhoff C, Mennel FJ et al (1996) Does fall in tissue glucose precede fall in blood glucose? Diabetologia 39:609–612
Gross TM, Bode BW, Einhorn D et al (2000) Performance evaluation of the MiniMed continuous glucose monitoring system during patient home use. Diabetes Technol Ther 2:49–56
Clarke WL, Cox D, Gonder-Frederick LA et al (1987) Evaluating clinical accuracy of systems for self-monitoring of blood glucose. Diabetes Care 10:622–628
Bolinder J, Kager L, Ostman J et al (1983) Differences at the receptor and postreceptor levels between human omental and subcutaneous adipose tissue in the action of insulin on lipolysis. Diabetes 32:117–123
Virtanen KA, Lonnroth P, Parkkola R et al (2002) Glucose uptake and perfusion in subcutaneous and visceral adipose tissue during insulin stimulation in nonobese and obese humans. J Clin Endocrinol Metab 87:3902–3910
Thome-Duret V, Reach G, Gangnerau MN et al (1996) Use of a subcutaneous glucose sensor to detect decreases in glucose concentration prior to observation in blood. Anal Chem 68:3822–3826
Aussedat B, Dupire-Angel M, Gifford R et al (2000) Interstitial glucose concentration and glycemia: implications for continuous subcutaneous glucose monitoring. Am J Physiol Endocrinol Metab 278:E716–E728
Caplin NJ, O’Leary P, Bulsara M et al (2003) Subcutaneous glucose sensor values closely parallel blood glucose during insulin-induced hypoglycaemia. Diabet Med 20:238–241
Monsod TP, Flanagan DE, Rife F et al (2002) Do sensor glucose levels accurately predict plasma glucose concentrations during hypoglycemia and hyperinsulinemia? Diabetes Care 25:889–893
Boyne MS, Silver DM, Kaplan J et al (2003) Timing of changes in interstitial and venous blood glucose measured with a continuous subcutaneous glucose sensor. Diabetes 52:2790–2794
Aman J, Berne C, Ewald U et al (1992) Cutaneous blood flow during a hypoglycaemic clamp in insulin-dependent diabetic patients and healthy subjects. Clin Sci (Lond) 82:615–618
Serne EH, IJzerman RG, Gans RO et al (2002) Direct evidence for insulin-induced capillary recruitment in skin of healthy subjects during physiological hyperinsulinemia. Diabetes 51:1515–1522
Aman J, Berne C, Ewald U et al (1990) Lack of cutaneous hyperemia in response to insulin-induced hypoglycemia in IDDM. Diabetes Care 13:1029–1033
Schmidtke DW, Freeland AC, Heller A et al (1998) Measurement and modeling of the transient difference between blood and subcutaneous glucose concentrations in the rat after injection of insulin. Proc Natl Acad Sci U S A 95:294–299
Acknowledgements
This work was supported by National Institutes of Health grants RO1-DK57210 (K. Rebrin), RO1-DK64701 (G. M. Steil) and M01-RR00865. We also wish to thank the nurses and staff of the UCLA General Clinical Research Center for help in performing the study.
Duality of interest
G. M. Steil and K. Rebrin are employed by Medtronic MiniMed, maker of the glucose sensor used here.
Author information
Authors and Affiliations
Corresponding author
Estimation of ISF glucose dynamics
Estimation of ISF glucose dynamics
Sensor current was analysed according to a two-compartment model of plasma and ISF glucose (Fig. 1). For the model, the mass balance equation characterising ISF glucose is:
where G 1 and G 2 are the plasma and ISF glucose concentrations respectively, K 12 and K 21 are the forward and reverse flux rates for glucose transport across the capillary, and V 1 and V 2 are the volumes of the plasma and ISF compartments. Glucose uptake into the s.c. tissue is characterised by K 02, which is expected to increase if insulin increases glucose uptake into the surrounding cells. Rewriting Eq. (1) in terms of identifiable parameters leads to:
where G b is the basal plasma glucose level. The plasma to ISF glucose gradient (G 2/G 1) is given by p 1/p 2 and the ISF glucose delay by 1/p 2 (p 1=K 21 V 1/V 2; p 2=(K 12+K 02)). For this formulation, delay time (1/p 2) is defined as the time to reach 63% equilibration.
For s.c. glucose sensing, sensor current (I sig) is proportional to the ISF glucose concentration:
where α is the intrinsic sensor sensitivity (nA·mmol−1·l). Substituting Eq. (3) into Eq. (2) leads to:
Here, p 3 is equal to the product of α times p 1. Parameter p 2 is unchanged from its definition in Eq. (2); thus, delay is dependent on the physiological parameters K 12 and K 02, but independent of the sensor’s intrinsic sensitivity (α). The plasma to ISF glucose gradient (p 1/p 2) is not identifiable, but changes in either α or p 1 alter the apparent sensor sensitivity (p 3/p 2), which is identifiable. Apparent sensitivity was also estimated from the steady-state ratio of I sig/G 1 at each clamp level, with steady state defined as the average of the last half-hour of each period.
To obtain estimates of the apparent sensor sensitivity (p 3/p 2) and ISF glucose delay (1/p 2), p 2 and p 3 were identified from Eq. (4) during the fall (0<t<270) and recovery (270<t<380 min) of plasma glucose. Identification was performed using non-linear least-squares routines available in Mlab (Civilized Software). The sensor current was well fitted by this procedure in 17 of 18 cases for both the fall and recovery in plasma glucose (r 2>0.8). In one case, the fit could not be obtained during the fall, and in a second case the fit could not be obtained during the recovery. In cases where the fit could not be obtained, the model parameters were treated as missing data but sensor profiles were retained for the purposes of calculating MAD, regression parameters and correlation (see Statistical analysis).
Rights and permissions
About this article
Cite this article
Steil, G.M., Rebrin, K., Hariri, F. et al. Interstitial fluid glucose dynamics during insulin-induced hypoglycaemia. Diabetologia 48, 1833–1840 (2005). https://doi.org/10.1007/s00125-005-1852-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-005-1852-x