
Abstract
Hypoglycemia is the most frequent complication of insulin therapy in patients with type 1 diabetes. Since the brain is reliant on circulating glucose as its main source of energy, hypoglycemia poses a threat for normal brain function. Paradoxically, although hypoglycemia commonly induces immediate decline in cognitive function, long-lasting changes in brain structure and cognitive function are uncommon in patients with type 1 diabetes. In fact, recurrent hypoglycemia initiates a process of habituation that suppresses hormonal responses to and impairs awareness of subsequent hypoglycemia, which has been attributed to adaptations in the brain. These observations sparked great scientific interest into the brain’s handling of glucose during (recurrent) hypoglycemia. Various neuroimaging techniques have been employed to study brain (glucose) metabolism, including PET, fMRI, MRS and ASL. This review discusses what is currently known about cerebral metabolism during hypoglycemia, and how findings obtained by functional and metabolic neuroimaging techniques contributed to this knowledge.
Similar content being viewed by others

Introduction
The brain is one of the most metabolically active organs in the body and it consumes energy disproportionate to its size. In humans, the brain represents only about 2 % of total body weight, yet it accounts for approximately 20 % of the body’s oxygen use and 25 % of the body’s use of glucose. Glucose is the primary fuel for the adult brain. In young adults, the ‘resting’ brain consumes approximately 110 g of glucose per day, i.e. 5.5 mg glucose per 100 g of brain tissue per minute [1]. Since the brain’s energy stores are small, normal brain function depends on a continuous supply of glucose from the bloodstream. Under normal conditions, the human body takes great effort and is very efficient in avoiding hypoglycemia in almost all circumstances to maintain sufficient glucose delivery to the brain.
Type 1 diabetes mellitus is an autoimmune-mediated disease, characterized by destruction of most, if not all, of the insulin-producing capacity of pancreatic beta-cells. As a consequence, supplemental insulin treatment is required to maintain glucose control and decrease the risk of complications resulting from hyperglycemia [2]. Unfortunately, therapeutic insulin is still poor at mimicking the pharmacology of endogenous insulin. As a consequence, people with type 1 diabetes—in particular those aiming for strict glycemic control—are at continuous risk of hypoglycemia, the average frequency of which has been estimated at two non-severe, symptomatic episodes per week [3–5] and one severe, potentially hazardous event, per year [4, 6, 7]. Although there is substantial variation in both the rate and the severity of hypoglycemia, both between and within persons [4, 8], this estimation means that the brains of people with type 1 diabetes are exposed to many thousands of hypoglycemic episodes over a lifetime of diabetes.
Studying brain metabolism during hypoglycemia may reveal the potential harmful effects of (recurrent) hypoglycemia on the brain and may increase our understanding of metabolic adaptations that might underlie impairments in the defenses against hypoglycemia [9]. Modern neuroimaging techniques have enabled the study of cerebral metabolism in vivo in a relatively non-invasive manner. This review will focus on the effect of hypoglycemia on brain (glucose) metabolism, with a particular emphasis on recent findings from functional and metabolic neuroimaging studies. Basic mechanisms of brain energy metabolism and neuroimaging techniques will be discussed briefly.
Glucose counterregulation
In healthy, non-diabetic humans, hypoglycemia is unlikely to ever occur due to a hierarchically coordinated system that integrates insulin secretion and counterregulatory hormone and symptom responses [10, 11]. When glucose levels in the low-physiological range (e.g. late post-absorptive or fasting state) tend to fall, insulin secretion is suppressed to such an extent that true hypoglycemia can almost always be prevented. When insulin is given to experimentally induce hypoglycemia in people without diabetes, glucose levels at or below ~3.8 mmol/L will induce a glucagon response, the secretion of which by pancreatic alpha-cells is probably controlled by the neighboring beta-cells [10, 11]. Such a glucose level also stimulates the secretion of adrenaline, whereas slightly lower levels are needed to elicit autonomic warning symptoms, such as sweating, palpitations, trembling and feeling hungry [10, 11]. These symptoms are aimed at initiating a behavioral response (i.e. ingesting carbohydrates). Further falls in plasma glucose values result in neuroglycopenic symptoms, which range from mild cognitive impairment, such as difficulty in concentrating, to overt confusion and even coma or seizures in its most severe form [10, 11].
In patients with type 1 diabetes, hypoglycemia typically results from the interplay between therapeutic peripheral hyperinsulinemia and impaired defenses against falling plasma glucose levels [10]. These impairments first include the inability to decrease insulin and to increase glucagon in response to hypoglycemia. The latter is not a structural defect, but specific for hypoglycemia and probably secondary to loss of control by non-functioning beta-cells [12]. In patients with longer diabetes duration and more frequent exposure to hypoglycemia, adrenaline responses to hypoglycemia become attenuated, in part due to a shift of these responses to lower glucose values [13]. The defective adrenaline responses are associated with, although not necessarily the cause of, similar defects in the emergence of autonomic symptom responses [14, 15]. Disappearance of these symptoms interferes with the ability to timely and accurately perceive, interpret and respond to falling plasma glucose levels. This inability is known as the clinical syndrome of impaired awareness of hypoglycemia and increases the risk of particularly severe hypoglycemia, defined as those events requiring assistance from another person [16], by a factor of six or more [17, 18]. Both the attenuated adrenaline response and impaired awareness of hypoglycemia are usually the result of (recurrent) antecedent hypoglycemia rather than of autonomic neuropathy, for which the term ‘hypoglycemia-associated autonomic failure’ (HAAF) has been introduced [19]. HAAF can be effectively treated by several weeks to months of scrupulous avoidance of hypoglycemia [15, 20, 21], although it appears that the symptomatic component responds earlier and better than the hormonal component [15]. The underlying mechanism(s) explaining the attenuating effect of prior hypoglycemia on responses to subsequent events have not been fully elucidated. However, there is agreement that alterations in the brain play a pivotal role.
Morbidity associated with hypoglycemia
The glucose level at which cognitive function declines is subject to substantial variation; in some people cognitive dysfunction already occurs at plasma glucose levels between 3.0 and 4.0 mmol/L, whereas others continue to function well at levels below 2.5 mmol/L [8, 22]. Almost all domains of cognitive function are potentially at risk during acute hypoglycemia, with complex tasks (e.g. car driving) being affected earlier than simple tasks [23, 24]. Prolonged and/or profoundly severe hypoglycemia may eventually cause neuronal death [25, 26]. The cerebral cortex and hippocampus are the most vulnerable regions in the brain to be injured by severe hypoglycemia, while the brain stem and cerebellum are most resistant [27]. However, although persistent vegetative states or brain death resulting from hypoglycemia have been described [28–30], most patients with type 1 diabetes recover uneventfully from even severe hypoglycemia complicated by seizures or coma, especially when they are young and in good clinical condition. In addition, evidence for an association between multiple episodes of severe hypoglycemia and long-term cognitive decline in people with type 1 diabetes is lacking [31, 32]. Finally, although 4–10 % of all deaths in patients with type 1 diabetes have been attributed to hypoglycemia, most of these deaths are thought to be either accidental (e.g. in traffic) or cardiovascular (e.g. arrhythmia) rather than the direct consequence of brain death [33, 34].
It should be noted that both the developing brain of young children with type 1 diabetes [35, 36] and the brain of the elderly, in particular in patients with type 2 diabetes [37, 38], seem more susceptible to harm from hypoglycemia. Children with type 1 diabetes performed worse on cognitive tests when they had a history of severe hypoglycemia below the age of 5 years, compared to patients without such a history and non-diabetic controls [35]. In prospective cohorts of people with type 2 diabetes, a history of severe hypoglycemia has been associated with cognitive decline and frank dementia [38], as well as with greater risks of cardiovascular events and death [39, 40]. On the cellular level, there are now indications that glucose deprivation may accelerate apoptosis of neurons, which could underlie neuronal cell death and predispose for cognitive decline [41]. It has also been speculated that the acute, physiological changes in hematological and hemodynamic parameters induced by hypoglycemia may be particularly damaging when the vasculature has already been injured [42, 43], possibly explaining the discrepancy between type 1 and type 2 diabetes [31, 44]. Another factor explaining this discrepancy may lie in the concept of hypoglycemic preconditioning. Rodents exposed to recurrent hypoglycemic events of moderate severity were less likely to develop neuronal damage or cognitive impairments or die during subsequent severe hypoglycemia than age-matched littermates who were not pre-exposed to hypoglycemia [45, 46]. These data may help to explain recent observations that patients with type 1 diabetes and impaired awareness of hypoglycemia, as a reflection of recurrent hypoglycemia, appeared not at greater risk of dying than patients with intact awareness [47].
The role of the brain in glucose counterregulation
The brain is not just at the receiving end of hypoglycemia, but it plays an important role in both the detection of hypoglycemia and in the subsequent initiation and coordination of counterregulatory responses to restore euglycemia, as described above. This system maintains glucose homeostasis through a classic sensory-motor integrative pathway in which a decrease in plasma glucose levels is detected by an extended network of glucose sensing neurons located within the brain and the periphery [48]. Specialized glucose-sensing cells are located in the hepatic portal/mesenteric vein, gut, carotid body and oral cavity. In the brain, glucose-sensing neurons are found at a number of locations, but particularly in the ventromedial nucleus of the hypothalamus (VMH) and areas that originate from the hindbrain. Integrative networks receive projections from these sensing neurons and subsequently assimilate their input with signals from other brain regions, such as information about circadian rhythm and arousal state. This information is relayed to motor neurons, which generate an output that drives the counterregulatory response and subsequently restores plasma glucose levels. Conversely, glucose sensing may influence other neural processes that have no role in glucose counterregulatory function [49].
Although the VMH is only one of a number of regions involved in the detection of hypoglycemia, it is thought to be the most important. The VMH serves as the central relay station for signals from many other regions and plays a crucial role in the coordination of the counterregulatory responses to falling glucose levels. Animal studies have shown that both destruction of the VMH and local perfusion of the VMH with glucose, disrupt counterregulatory hormone responses to systemic hypoglycemia [50, 51]. Conversely, local glucopenia in the VMH stimulates these responses in the absence of hypoglycemia [52]. In analogy, glucose counterregulation was also found defective in a patient with lesions from sarcoidosis in the hypothalamus, presumably due to destruction of the glucose-sensing neurons in the VMH [53].
The mechanism of glucose sensing by the brain, in particular in the VMH, has not been fully clarified. Two main types of glucose-sensing neurons have been identified: glucose-excited neurons, whose activity increases as glucose levels rise, and glucose-inhibited neurons, which become more active as glucose levels fall and less active when they rise [54]. These neurons ‘sense’ a fall in glucose probably as a result of alterations in ATP/ADP and AMP/ATP ratios, respectively, following a reduction in glucose metabolism. This could explain why fuelling the VMH with an alternative source of energy, such as lactate, suppresses glucose counterregulation [55, 56]. The subsequent intracellular actions that may ultimately lead to a counterregulatory response probably involve activation of AMP-activated protein kinase, formation of nitric oxide and release of glutamate in glucose-inhibited neurons. Other potential mediators involved in these responses include (a decrease in) gamma-aminobutyric acid (GABA) release from glucose-excited neurons, noradrenaline, serotonin and corticotrophin-releasing hormone [57]. For further reading on this subject, we refer to recent reviews by McCrimmon [54] and Chan and Sherwin [57].
Cerebral glucose delivery, uptake and metabolism
Glucose is transported across the blood–brain barrier into extracellular fluid (ECF) by facilitated diffusion, mediated via glucose transporter protein 1 (GLUT1) [58]. The predominant transporters involved in subsequent glucose uptake from the ECF in neurons and in astrocytes are GLUT3 and GLUT1, respectively [59], both insulin-independent glucose transporters. Once intracellular, glucose is phosphorylated by hexokinase as the initial step of glucose metabolism. The glucose-6-phosphate (Glc-6-P) thus produced can enter several metabolic pathways in the brain [60].
In 1945, Kety and Schmidt developed the first method to quantitatively assess brain glucose uptake in humans in vivo and to derive data on its subsequent metabolism [61]. This highly invasive technique required the use of arterial and internal jugular vein catheterizations to determine arteriovenous concentration differences for glucose, which together with measurement of global cerebral blood flow (CBF) were then used to calculate the global cerebral metabolic rate of glucose [61–64]. In humans, the Kety–Schmidt method was used to show that brain glucose uptake falls during hypoglycemia and that this coincides with the appearance of counterregulatory hormone responses and autonomic warning symptoms [65, 66]. However, whether these data can be used to reliably assess brain glucose metabolism is a matter of debate. Indeed, the calculations rely solely on the disappearance rate of glucose from the circulation. Therefore, this technique cannot discriminate between specific metabolic steps and ignores the potential contribution of other metabolites. Moreover, the highly invasive nature of the Kety–Schmidt technique is a considerable limitation for research in humans.
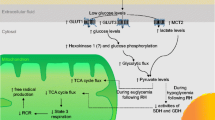
The past 40 years have shown rapid advances in modern metabolic and functional neuroimaging techniques to study brain (glucose) metabolism vis-à-vis CBF during hypoglycemia, including positron emission tomography (PET), functional magnetic resonance imaging (fMRI) and magnetic resonance spectroscopy (MRS). It is important to note that the distribution of CBF and the cerebral metabolic rate of glucose (CMRglc) are closely linked to local brain activity. Brain activation causes proportionate increases in both local CBF and CMRglc. These processes are being referred to as neurovascular coupling or neurometabolic coupling, respectively, and hypothesized to be mediated by neurotransmitter release and vasoactive metabolic products [67]. Many functional neuroimaging techniques, including fMRI, rely on neurovascular coupling. The principles of the various imaging techniques will be briefly discussed (Fig. 1).

A simplified illustration of the multiple metabolic pathways of glucose in the brain and the metabolic signals used in different neuroimaging techniques. The initial step of glucose metabolism is phosphorylation of glucose to glucose-6-phosphate (Glc-6-P) by hexokinase. Glc-6-P can enter several metabolic pathways in the brain. It can be metabolized to produce energy via glycolysis or the TCA cycle. Glycolytic and TCA cycle intermediates are also used for the synthesis of amino acids and neurotransmitters. In addition, Glc-6-P is a precursor for glycogen. Lastly, metabolism of Glc-6-P via the pentose phosphate pathway (PPP) provides pentose for nucleotide synthesis and NADPH, required for reductive reactions, such as lipid synthesis and for protection against oxidative stress. Arteriovenous concentration differences (AV dif) can be used to estimate global cerebral metabolic rate from the disappearance of metabolites from the circulation. PET (depicted in orange) uses radiolabeled glucose analogues (such as FDG), which are trapped early in metabolism (for example fluorodeoxyglucose-6-phosphate/FDG-6-P), to estimate rates of glucose uptake and metabolism. 31P MRS (depicted in blue) provides information about ATP production and thus brain energy metabolism. 13C-MRS (depicted in green) is useful for estimating TCA cycle fluxes and CMRglc, derived from 13C label incorporation into specific metabolites (Glu, Gln). Both ASL and BOLD fMRI provide estimates of CBF
PET
Positron emission tomography can be used to measure emissions from a variety of radioactively labeled tracers in the brain to quantify CBF, glucose uptake and phosphorylation, oxygen consumption and brain receptors for major neurotransmitters, depending on the type of radiotracer used [68]. 15O-labeled water PET, for example, has been commonly applied to quantify regional CBF [69, 70]. For the study of brain glucose metabolism, [18F]fluoro-2-deoxy-d-glucose (FDG) is the most widely used tracer. FDG is taken up by the brain in a similar manner as glucose, but unlike native glucose, once phosphorylated (FDG-6-P), it cannot be metabolized further, resulting in accumulation of the tracer in the cell. Under steadystate conditions, in which total influx of metabolites into a pathway equals the outflow, the rate of tracer accumulation in the brain can be used to estimate global and regional rates of glucose transport and metabolism [58]. PET has been particularly valuable in studying the effect of hypoglycemia on CBF, brain glucose uptake and cerebral metabolic rate in humans with and without type 1 diabetes [71–74]. However, this technique cannot be used to study glucose metabolism downstream of its conversion to glucose-6-phosphate [75]. Also, animal studies suggest that the lumped constant, a correction factor that relates the metabolic rate of FDG to that of native glucose [76], may increase during hypoglycemia [77, 78]. The tracer [11C]3-O-methyl-d-glucose (3-OMG) may provide more robust information about cerebral glucose uptake at varying glucose concentrations, as it is not phosphorylated [79], but its relative short half-life time (~20 min) and complex preparation limits the use of this compound in a clinical setting [80].
fMRI
Functional magnetic resonance imaging is primarily used to study regional neuronal activation (patterns) by the detection of changes in oxygen demand by the brain [81], based on the concept of neurovascular coupling described above. Blood oxygenation level dependent (BOLD) contrast is one of the primary contrast mechanisms for fMRI, which exploits the differences in magnetic properties between deoxygenated and oxygenated hemoglobin [82]. Regional brain activation increases local oxygen demands, but because the consequent increase in CBF exceeds these demands, the balance between deoxygenated and oxygenated hemoglobin changes towards the latter. This change in hemoglobin oxygenation can be probed and detected, so that a brain map of regions with increased or decreased activation can be constructed [81]. fMRI has been especially useful in detecting brain activation patterns in response to specific cognitive tasks or visual stimulation. Hypoglycemia has been reported to reduce regional BOLD activation in response to these tasks, but less so in patients with type 1 diabetes [83] than in non-diabetic subjects [84, 85]. These reductions in BOLD responses are commonly attributed to decreased neuronal activity, yet the potential impact of hypoglycemia on (global or regional) CBF, neurovascular coupling or oxidative metabolism, remains to be determined.
ASL
Arterial spin labeling (ASL) is an MRI method that provides non-invasive quantification of global and regional CBF. ASL does not require an exogenous contrast agent, but uses magnetically labeled arterial blood water as a diffusible tracer. Arterial blood water is first labeled magnetically using radiofrequency (RF) pulses. Subsequently, this labeled arterial blood flows into the brain where it exchanges with tissue water, after which an image is taken. The experiment is then repeated without labeling the arterial blood to create a control image. The signal difference between control and labeled images reflects local CBF [86, 87]. While the signal-to-noise (SNR) ratio in BOLD fMRI is higher, ASL measures brain perfusion more directly, enables quantification of CBF, and is suitable for studying variations in CBF over a longer period of time due to stable noise characteristics [88]. ASL thus allows the detection of changes in CBF during hypoglycemia and is, in contrast to fMRI, less dependent on other metabolic parameters, such as oxygenation or glucose concentrations that might change during hypoglycemia. A high magnetic field (e.g. 3 T) is usually recommended to improve SNR when performing ASL.
MRS
Magnetic resonance spectroscopy is a non-invasive technique, closely related to MRI. Both techniques make use of the spin properties of certain nuclei when brought into a magnetic field. For MRI, the proton nucleus is used to construct a highly detailed anatomical image based on the different water concentrations in various tissues. For MRS, these spin properties are used to determine the concentration of specific metabolites in the tissue examined. These concentrations are derived from the peaks in a spectrum [89]. MRS is feasible on any nucleus possessing a magnetic moment, but is most frequently performed on the high natural abundant and MR sensitive proton nucleus (1H), providing steady-state information on concentrations of proton-containing brain metabolites at a single time point [90]. However, because water contains most of the proton nuclei, the water signal needs to be suppressed to allow reliable measurements of metabolite concentrations. As a consequence, the sensitivity of MRS is manifold lower than that of MRI, even at high magnetic fields. Nevertheless, since nearly all metabolites contain protons, 1H-MRS is a powerful technique to identify and quantify a large number of metabolites relevant for glucose metabolism (e.g. lactate, glutamate, glutamine) at in vivo concentrations typically above 0.5 mM.
The use of carbon-13 (13C) in MRS is specifically relevant for the study of brain glucose metabolism. Carbon exists in the human body in two isotopes, of which carbon-12 (12C) is dominant with a natural abundance of 98.9 %. 12C does not possess a net nuclear spin and consequently cannot be detected by MRS. In contrast, 13C does possess a magnetic moment, but has a very low natural abundance of 1.1 %. However, the intravenous infusion of 13C-enriched substrates, such as [1-13C]glucose, [3-13C]lactate or [3-13C]acetate, offers the possibility to study fluxes of these substrates in the brain through important metabolic pathways (Fig. 2).

Time series of 13C-MR spectra, acquired from a ~125 mL voxel, placed in the occipital cortex. Spectra are averaged over 20 min, after administration of [1-13C]glucose during a hypoglycemic clamp in one healthy subject. Once the infused [1-13C]glucose is taken up by the brain and incorporated into various glucose metabolites, an increase in signal over time is observed. Numbers indicate the position of the 13C label, as explained in more detail in Fig. 3. Asp aspartate, Gln glutamine, Glu glutamate, Lac lactate (from Ref. [143], with permission from Elsevier)
For the study of brain glucose metabolism, it is important to note that [1-13C]glucose is taken up and metabolized by the brain similar to native (i.e. unlabeled) glucose. Following transport across the blood–brain barrier and phosphorylation by hexokinase, glucose is the main substrate for the production of energy (by formation of ATP) via glycolysis and the tricarboxylic acid (TCA) cycle [82]. As such, 13C-MRS allows the fate of the 13C-labeled glucose to be followed as it flows into glycolysis. The 13C-label is transferred from glucose on the C-1 position to pyruvate on the C-3 position during glycolysis and, subsequently, passes through all metabolites of the TCA cycle. In this process, the 13C-label is incorporated into the MRS-detectable metabolites glutamate, glutamine and aspartate, all at specific carbon positions. Because these positions change during the second time the isotope flows in the TCA cycle (Fig. 3), the time-course of 13C-label incorporation into these metabolites can be used as input for a metabolic model to calculate the TCA cycle flux and CMRglc [91]. However, although the fates of individual carbon atoms can be tracked in the TCA cycle, cerebral 13C-MRS provides no information about the loss of label in diffusible metabolites, such as glutamine and lactate which may exchange between brain and blood plasma [82, 92].

One-compartment metabolic model describing the incorporation of 13C label from (infused) [1-13C]glucose into the TCA cycle and its metabolites. When taken up by the brain, the 13C-label is first incorporated into the C3 position of pyruvate and subsequently into the C3 position of lactate. Once the 13C-label continues through the TCA cycle, it is incorporated into the C4 position of αKG, glutamate and glutamine. In the second turn of the cycle, the label is equally distributed over the C2 and C3 positions of these metabolites. The TCA cycle flux can be estimated using a metabolic model where the time courses of the uptake of the 13C-label in glutamate and glutamine in the different carbon positions, measured with 13C-MRS, are used as input. Filled circles represent the carbon position that is labeled with 13C, white circles represent unlabeled carbons. αKG α-ketoglutarate, BBB blood–brain-barrier, Glc glucose, Gln glutamine, Glu glutamate, Lac lactate, LDH lactate dehydrogenase, Pyr pyruvate, V gln exchange rate between glutamate and glutamine, V TCA TCA cycle rate, V x exchange rate between α-ketoglutarate and glutamate
13C-MRS has been proven to be a valuable imaging technique to study brain (glucose) metabolism via specific pathways in humans in vivo, under various conditions, including hyperglycemia [93] and hypoglycemia [94]. In addition, 13C labeled compounds other than glucose, such as 13C-acetate and 13C-lactate, can be used to provide a more complete picture of the very complex metabolic processes in the brain. Indeed, 13C-acetate, which is metabolized almost exclusively in astroglia [95], has been used to distinguish astroglial and neuronal metabolism more directly, and to study transport and metabolism of non-glucose fuels during hypoglycemia [96, 97].
Phosphorus-31 (31P) is another naturally abundant nucleus with a relatively high sensitivity for MRS. 31P-MRS of the brain can be used to detect metabolites that play a key role in brain energy metabolism and provides information on flux through the creatine kinase reaction (e.g. ATP, phosphocreatine, inorganic phosphate), intracellular pH and magnesium concentrations [98]. Thus far, this technique has been seldom used to study brain metabolism during hypoglycemia [99].
Cerebral nutrient transport capacity and hypoglycemia
Glucose uptake
As mentioned above, glucose uptake into the brain occurs through facilitated transport independent of insulin. As a consequence, there is a linear relationship between plasma glucose concentrations and brain glucose content over a range of plasma glucose values up to ~30 mmol/L [100–103]. This linear relationship also extends well into the hypoglycemic range, although data below plasma levels of ~2.5 mmol/L are missing in humans (Fig. 4) [104]. To explain HAAF, it has been hypothesized that chronic or repeated hypoglycemia increases glucose transport capacity over the blood–brain barrier to compensate for the fall in glucose availability to the brain during subsequent hypoglycemia. Indeed, several animal studies have shown that days to weeks of chronic hypoglycemia cause upregulation of brain glucose transporters, including both GLUT-3 on neuronal membranes [105, 106], and GLUT-1 on the vascular endothelium at the blood–brain barrier [107, 108]. In accordance, Boyle and co-workers applied the Kety and Schmidt technique to show preservation of brain glucose transport rather than a fall during hypoglycemia in healthy volunteers after prior exposure to hypoglycemia, whereas it fell when such exposure had not taken place [65]. The investigators went on to report similar findings of preserved glucose transport in patients with type 1 diabetes and near-normal glycosylated hemoglobin (HbA1c), possibly reflecting high hypoglycemic burden, as they also had reduced awareness of hypoglycemia [66].

Linear relationship between plasma and brain glucose levels under normo- and hypoglycemic conditions in healthy subjects (open squares) and patients with type 1 diabetes (closed circles). Brain glucose levels were measured with 13C-MRS. The plasma versus brain glucose relation was fitted with linear regression analysis to determine reversible Michaelis–Menten kinetics to show the best fit of the data with 95 % confidence intervals. R 2 = 0.59, P < 0.001. Assuming continuation of this linear relationship between plasma and brain glucose levels, brain glucose approaches zero at a plasma glucose level of approximately 1.2 mmol/L (from Ref. [104], with permission from the American Diabetes Association)
In mice and in rats, very low plasma glucose values, typically well below 2.0 mmol/L, have been found to proportionally increase brain glucose uptake as a function of increased cerebral perfusion [102, 109]. Various neuroimaging studies investigating glucose transport over the blood–brain barrier in humans have produced conflicting results. A 1H-MRS study performed under hyperglycemic conditions showed greater brain glucose concentrations in patients with type 1 diabetes and impaired awareness of hypoglycemia than in people without diabetes [110]. However, a similar study found no evidence of altered brain glucose transport in healthy volunteers subjected to antecedent repeated hypoglycemia, despite clearly attenuated hormone responses to hypoglycemia [111]. In accordance, global blood-to-brain glucose transport, as measured with [1-11C]-glucose PET, remained unaltered in healthy volunteers after exposure to 24 h of moderate hypoglycemia, albeit interspaced with transient glucose normalizations during meals [112]. Finally, a 3-OMG-PET study also showed no differences in global brain glucose transport during hypoglycemia between patients with normal and those with impaired awareness of hypoglycemia [72].
Monocarboxylic acid (MCA) uptake
Although glucose is its principal source of energy, the brain may resort to alternative non-glucose fuel substrates under glucopenic conditions. These alternative substrates include foremost ketones, and lactate, which enter the TCA cycle after conversion to pyruvate or acetyl coenzyme A, and can be metabolized in a similar way as glucose to sustain brain metabolism, and spare glucose.
Ketones such as beta-hydroxybutyrate and acetoacetate are synthesized in the liver from fatty acids during prolonged fasting, starvation and severe carbohydrate restriction. Under such conditions, up to 60 % of brain energy requirements may be derived from ketone metabolism [113], whereas ketogenic diets can more or less restore brain energy metabolism and prevent epileptic seizures in patients with GLUT1 deficiency who are unable to transport glucose into the brain [114, 115]. However, because insulin suppresses the production of ketones, the brain is usually unable to use this source of energy during insulin-induced hypoglycemia [116]. PET studies with the use of both ketone and glucose tracers may help to unravel the complex interaction between the metabolism of ketones and glucose by the brain under different circumstances, including hypoglycemia [113].
In recent years, it has gradually been recognized that lactate plays an important role in the energy metabolism in the brain, particularly during hypoglycemia since both hypoglycemia and insulin increase plasma levels of lactate, at least in healthy subjects [94, 117, 118]. Under basal, euglycemic, conditions, the contribution of systemic lactate to cerebral energy metabolism is approximately 8–10 %. However, the proportional contribution of lactate has been reported to increase during strenuous exercise, when plasma lactate levels rise substantially [119, 120]. The role of lactate in specific areas of the brain includes its involvement in or interference with hypoglycemia detection in the VMH as stated above. Lactate has also been found to be a crucially monitored variable in the detection of energy imbalance in the caudal hindbrain [121]. The importance of lactate for the brain was first highlighted when Pellerin and Magistretti published their astrocyte-neuron lactate shuttle (ANLS) hypothesis. This hypothesis posits that glucose is taken up by and metabolized in astrocytes to form lactate, after which lactate is exported to neighboring neurons where it is oxidized, especially during activation [122]. This concept, which bears analogy to the cell–cell lactate shuttle, through which skeletal muscle can transport a non-glucose energy source to other organs [123], thus suggests that astrocytes play the primary role in brain glucose metabolism. Simpson et al. later came to a different conclusion and developed a model that basically adopts the opposite view, in which neurons are the principal site of glucose uptake and metabolism, and the chief exporter of lactate. This hypothesis was therefore termed the neuron-astrocyte lactate shuttle (NALS) [59] and fuelled a heavy debate [124, 125]. The debate on the direction of the lactate shuttle is ongoing with studies identifying the neuron as the principal locus of glucose uptake [126], and other studies indicating that neurons rather than astrocytes are the primary sites for oxidation of exogenous lactate [119, 127].
Monocarboxylic acid transporters (MCTs) facilitate the uptake of lactate as well as that of acetate and ketone bodies into the brain, the expression of which may increase following sustained hyperketonemia or recurrent hypoglycemia. A recent study in rats demonstrated a twofold increase in the expression of MCTs 1 and 2 in the cerebral cortex after the induction of diabetes by streptozotocin. After 8 weeks of frequent, prolonged endurance training and concomitant exposure to hypoglycemia after and between exercise sessions, the expression of both transporters increased even further [128]. Such greater transport capacity may explain recent observations in which recurrent exposure to hypoglycemia increased the uptake of 13C-labeled lactate into the rat brain under hypoglycemic conditions [129]. During hypoglycemia, the uptake of both acetate and lactate into the human brain, as measured by 13C-MRS during infusion of 13C-labeled acetate or lactate, respectively, was found to be considerably greater in patients with well-controlled type 1 diabetes than in healthy controls [97, 130].
Transport and uptake of other substrates
Oral intake of amino acids has been reported to enhance the glucagon response to hypoglycemia and to improve some aspects of cognitive function during hypoglycemia in non-diabetic and diabetic subjects [131, 132]. Amino acids might also serve as a non-glucose substrate that could be used by the brain as an alternative fuel and to sustain cognitive function during hypoglycemia. Early studies showing utilization of amino acids by the rat brain during prolonged hypoglycemia and of amino acids contributing to glycogen synthesis in brain cell cultures supported this theory [133, 134]. However, data obtained in humans using arteriovenous concentration differences found no evidence that greater availability of amino acids increased its net brain uptake during hypoglycemia [135] or was able to offset energy deficit due to reduced glucose supply [136].
A few studies have investigated whether the human brain can use lipid substrates to support cerebral metabolism and brain function during hypoglycemia. Fatty acids can readily cross the blood–brain barrier to be oxidized by the brain, as demonstrated by a 13C-MRS study in rats [137]. In healthy humans, elevated plasma levels of non-esterified fatty acids and glycerol were found to reduce hormonal and symptom responses to hypoglycemia, but could not protect against the fall in cognitive function [138]. Conversely, in a more recent study, ingestion of medium-chain triglycerides maintained cognitive function during hypoglycemia without affecting adrenergic or symptomatic responses to hypoglycemia in intensively treated subjects with type 1 diabetes [139]. It should be acknowledged, however, that the inferences made with respect to the uptake of lipid substrates in the brain were indirect and that no neuroimaging studies have been performed that evaluated the effects of these substances on cerebral metabolism more directly.
Brain metabolism during hypoglycemia
Glucose metabolism
Both PET and MRS have been used to investigate the effect of hypoglycemia on brain glucose metabolism. PET has been particularly useful in detecting regional differences in tracer accumulation in the brain, both during hypoglycemia [71], and after restoration to euglycemia [74]. However, rather than focusing on glucose uptake or metabolism, the close link with neuronal activation is then exploited to use the data as input factors for mapping regional brain activity. Thus, the observation that CMRglc relatively increased during hypoglycemia in patients with type 1 diabetes and normal awareness of hypoglycemia, and relatively fell in patients with impaired awareness of hypoglycemia, was interpreted as an increase in brain activation and absence of such a response [72]. When this increased activation would occur in brain areas involved in the perception of and the generation of responses to hypoglycemia, the lack of increased activation could then underlie loss of hypoglycemic awareness [72, 140]. Support for this hypothesis came from another FDG-PET study [141] and a subsequent analysis of these data [73], as tracer uptake in areas that engage appetite control and food-seeking behavior was reduced in patients with impaired compared to patients with intact awareness of hypoglycemia.
As outlined above, 13C-MRS in combination with infusion of 13C-labeled glucose has the unique property that it enables the investigation of cerebral glucose metabolism in humans in vivo. Since the SNR is relatively low, most studies employing this technique used large doses of isotopically enriched glucose at high enrichment percentages. Measurements have consequently generally been performed under hyperglycemic conditions with glucose levels up to 17 mmol/L and plasma C-13 enrichment values exceeding 60 %. Under such conditions, Henry et al. [93] reported no differences in the TCA cycle rate between patients with type 1 diabetes with impaired awareness of hypoglycemia and healthy controls. More recently, an improved sensitivity of the 13C-MRS method [142] in combination with an optimized 13C-glucose infusion protocol enabled us to study glucose metabolism in the human brain during hypoglycemia at lower enrichment values [143]. With this optimized technique, no differences were observed in cerebral glucose metabolism between hypoglycemia and euglycemia, neither in healthy controls [94], nor in patients with type 1 diabetes [144]. Under hypoglycemic conditions, however, the TCA cycle rate was approximately 45 % higher in patients than in healthy subjects, and inversely related to HbA1c. Appreciating a low HbA1c as a proxy for a high hypoglycemic burden, these data suggested a role for prior hypoglycemic exposure in the higher TCA cycle rate in patients with type 1 diabetes. Differences in brain glucose levels did not explain the preservation of brain metabolism and the higher TCA cycle rate in the patients, which suggested influx of a non-glucose carbohydrate source [104]. In an animal study by Herzog et al. [129], brain glucose transport capacity during hypoglycemia became rate limiting for TCA cycle activity in control animals, but not in rats exposed to antecedent recurrent hypoglycemia. Explanations for the discrepancy between the human and rodent data include the different species and the fact that the hypoglycemic condition was more profound in the animals. Indeed, studies in mice suggest that intracellular brain glucose concentrations approach depletion at plasma glucose values between 2 and 3 mmol/L [109].
Glycogen metabolism
The brain is able to store glycogen and to use this compound when plasma glucose levels are low, although its capacity to do so is very limited compared to other tissues such as skeletal muscle and the liver. It was long assumed that this presence of glycogen was restricted to astrocytes. However, a recent study showed that neurons contain a low but measurable amount of glycogen, the use of which was found to protect against hypoxic stress, at least in neuronal cell cultures and animal models [145]. Both in rodents [146] and in humans [147], it was shown that brain glycogen was used during hypoglycemia, and that its stores were replenished above baseline levels after restoration of euglycemia, a phenomenon termed glycogen supercompensation. It has been speculated that this expanded source of glucose within the brain could contribute to the development of impaired awareness of hypoglycemia by fuelling the brain or at least those areas involved in glucose-sensing during subsequent hypoglycemia [146]. However, prior exposure to recurrent hypoglycemia neither facilitated nor impaired access to glucose from glycogen in the rat brain during subsequent hypoglycemia [148]. Additionally, brain glycogen content, as measured by 13C-MRS in conjunction with 13C-glucose administration, was lower rather than higher in patients with type 1 diabetes and hypoglycemia unawareness [149].
Glutamate metabolism
Glutamate is the major excitatory neurotransmitter in the brain, but has many other metabolic fates, including the formation of glutamine, GABA and glutathione [150]. In addition, a new concept has been introduced by Sonnewald [151], who proposed that glutamate degradation in astrocytes contributes to most of the lactate that is released from the brain under resting conditions, offering a novel explanation for the concept of aerobic glycolysis in the resting state [151, 152]. Lastly, glutamate can be oxidized for the production of energy [150]. To this end, glutamate production in the brain is tightly coupled to TCA cycle activity [153]. Using 1H-MRS, Bischof et al. reported that hypoglycemia reduced the cerebral glutamate to creatine ratio in healthy controls, but not in patients with type 1 diabetes [154]. Similar results were reported by a more recent 1H-MRS study, were hypoglycemia reduced brain glutamate levels in healthy controls and in patients with type 1 diabetes with normal hypoglycemic awareness, but not in patients with impaired awareness of hypoglycemia [155]. The authors concluded that the preservation of brain glutamate during hypoglycemia in the latter group reflected a metabolic adaptation that eliminated the need to oxidize glutamate. They speculated that this adaptation could be augmented transport of glucose or of alternative fuels to the brain.
Metabolism of monocarboxylic acids
As discussed earlier, MRS studies using 13C-labelled acetate and lactate have clearly suggested that the capacity to transport MCAs over the blood–brain barrier during hypoglycemia is increased in patients with well-controlled type 1 diabetes. Indeed, a study during which 13C-acetate was infused under hypoglycemic conditions showed more than twofold higher brain acetate concentrations in subjects with type 1 diabetes compared to healthy controls. This greater acetate availability translated into a fraction of oxidative metabolism that resulted from acetate to be similarly increased [97]. In accordance, the relative contribution of acetate to brain metabolism in rats exposed to recurrent antecedent hypoglycemia was also increased during next-day hypoglycemia, indicating that brain substrate preferences may change rapidly from glucose to alternative substrates if needed [156]. To delineate whether this effect was a function of diabetes, prior hypoglycemia or both, the investigators repeated their 13C-acetate study in patients with type 1 diabetes with normal or impaired awareness of hypoglycemia and in healthy controls. They found that absolute rates of acetate metabolism during hypoglycemia were only higher in the patients with impaired awareness of hypoglycemic, suggesting that changes in acetate metabolism are the consequence of prior exposure to hypoglycemia rather than of diabetes per se [96].
Lactate uses the same MCT as acetate to cross the blood–brain barrier. Since plasma levels of lactate are approximately tenfold higher than those of acetate, and hypoglycemia stimulates the production of lactate [117], it seems plausible that lactate is the more likely substrate for brain metabolism when glucose levels are low. Studies dating back to the 1990s have shown that exogenous administration of lactate attenuates counterregulatory responses to and preserves cognitive function during hypoglycemia, presumably because lactate is used as an alternative source of energy by the brain [157–160]. In agreement, brain lactate concentrations during hypoglycemia, derived from the cerebral uptake of 13C-labelled lactate, were several fold higher in patients with type 1 diabetes with a history of frequent hypoglycemic episodes than in non-diabetic subjects [130] and in rats exposed to recurrent hypoglycemia versus those not exposed [129]. Surprisingly, the authors found no indication of greater lactate oxidation, as reflected by unchanged 13C fractional enrichments of brain glutamate and glutamine [130]. Data from the rodent study, in which lower glucose levels were achieved than in the human study, suggested that prior hypoglycemic exposure increased both the uptake and the oxidation of glucose by the brain, despite the higher lactate levels [129]. However, when the animal brain was stimulated during hypoglycemia, animals exposed to recurrent hypoglycemia had a partial loss of their functional cortical response, which was only normalized after the administration of lactate. This suggests that the higher capacity for lactate transport only becomes critical when the brain is activated during (deep) hypoglycemia.
Cerebral blood flow and hypoglycemia
There is uncertainty as to whether hypoglycemia affects global CBF and in what direction. Previous research in both patients with type 1 diabetes and healthy controls has reported either no change in global CBF during hypoglycemia [65, 66, 136], a modest increase [161–163], or even a slight decrease [71]. Differences in the plasma glucose levels achieved during hypoglycemia and, more importantly, in imaging techniques probably explain many of the discrepancies. Studies that investigated the effect of hypoglycemia on regional relative changes in CBF seem to have produced more consistent data. Both in healthy controls [161] and in patients with type 1 diabetes [164], hypoglycemia was found to increase blood flow to the frontal lobes. This relative redistribution of regional CBF was already observed under euglycemic conditions in patients with type 1 diabetes, and was more pronounced in patients who had experienced frequent hypoglycemia [165]. Since the frontal lobes are among the most vulnerable brain areas to suffer structural damage, this may be an adaptive response to prevent such damage by maintaining fuel supply during subsequent hypoglycemia.
Hypoglycemia has also been found to increase CBF in the thalamus [71, 166, 167] and hypothalamus [168, 169]. Mild or moderate hypoglycemia caused a rise in CBF in the hypothalamus in healthy non-diabetic subjects, as assessed by fMRI [168] or ASL [169], which preceded the rise in counterregulatory hormone responses seen during hypoglycemia [169]. Interestingly, Mangia et al. found blunting of this increase in thalamic perfusion during hypoglycemia in patients with type 1 diabetes with hypoglycemia unawareness, and a correlation between thalamic perfusion and the adrenaline response to hypoglycemia [167]. In contrast, recurrent hypoglycemia enhanced, rather than decreased, thalamic perfusion during subsequent hypoglycemia in healthy controls [170], so that the role of this brain region in the adaptation to hypoglycemia remains uncertain.
Discussion
The scientific field of metabolic and functional neuroimaging techniques for the brain has tremendously progressed over the past couple of decades. The application of these techniques to hypoglycemia research has considerably advanced our understanding of the brain’s responses to hypoglycemia. As plasma glucose falls below levels that can be reversed by responses at the level of pancreatic islets, i.e. suppression of insulin release and stimulation of that of glucagon, the brain’s sensing abilities are activated to allow timely detection of hypoglycemia. Data from functional and metabolic neuroimaging techniques now suggest that such moderate hypoglycemia neither affects the perfusion to nor the uptake of glucose into the brain, at least not globally, unless much deeper levels of glucose are achieved [102, 109]. In accordance, cerebral glucose metabolism appears largely maintained during moderate hypoglycemia [94, 129, 144]. However, on the regional level, moderate hypoglycemia causes redistribution of CBF to various brain areas involved in the detection of hypoglycemia, particularly the (hypo)thalamus [71, 166–169], where enhanced neuronal activation stimulates glucose uptake and metabolism. Such enhanced neuronal activation has also been found to occur in brain areas involved in appetitive motivational networks [73], thus linking the detection of hypoglycemia to a behavioral response.
Modern neuroimaging studies have revealed that recurrent hypoglycemia, which typically affects people with type 1 diabetes and underlies the clinical syndrome of impaired awareness of hypoglycemia, may initiate cerebral adaptations at many different levels. First, there is interference with the accurate detection of hypoglycemia, probably occurring at the level of the VMH. Brain areas that control appetite and induce fear and anxiety may not become activated during hypoglycemia. Whether locally increased glucose uptake in or reduced neuronal activation of the hypothalamic area (or both) form the underlying mechanism remains to be revealed. Importantly, it should be acknowledged that neurovascular coupling may be altered as a consequence of diabetes per se [171], chronic hyperglycemia [172] or microangiopathy [173], thus limiting the interpretation of studies relying on this concept. There is conflicting evidence as to whether recurrent hypoglycemia can stimulate brain glucose uptake during hypoglycemia [65, 66], although most studies employing neuroimaging techniques found no evidence for this suggestion [72, 111, 112]. Nevertheless, patients with type 1 diabetes, particularly those with impaired awareness of hypoglycemia [155], seem better able in maintaining brain (glucose) metabolism during hypoglycemia than healthy controls [94, 144, 154, 155], probably as a consequence of prior hypoglycemia [144]. Since profound hypoglycemia will eventually cause brain glucose metabolism to deteriorate [129], such an adaptation may shift the threshold for deterioration of the metabolic rate to lower plasma glucose levels.
Several mechanisms have been proposed that could explain the discrepancy between hypoglycemia-induced preservation of cerebral glucose metabolism and the fall in glucose availability during hypoglycemia. It seems likely that influx of a non-glucose energy substrate plays a role. Recent (neuroimaging) studies found little evidence to support enhanced blood to brain transport of amino acids [135, 136] or lipid substrates transport [138, 139] and ketones are unlikely candidates because its production is suppressed by insulin. Also, the lower brain glycogen content in patients with type 1 diabetes with impaired awareness of hypoglycemia compared to controls [149] argues strongly against the glycogen supercompensation hypothesis. Several arguments suggest a major role for lactate in preserving brain glucose metabolism during hypoglycemia. These include: (1) lactate can be used by the brain and may even be preferred over glucose under non-hypoglycemic conditions [174, 175]; (2) the capacity for lactate transport over the blood–brain barrier is increased in patients with impaired awareness of hypoglycemia and in rats after exposure to hypoglycemia [97, 129]; (3) the use of lactate by glucose-sensing neurons in the VMH may interfere with hypoglycemia sensing [54, 56, 57]. However, there are data that suggest that lactate is not used as major energy source for the brain during moderate hypoglycemia, despite greater availability [129, 130]. Also, it is not yet known whether brain uptake or metabolism of endogenously produced lactate is increased during hypoglycemia in patients with type 1 diabetes and impaired awareness of hypoglycemia. Finally, it has been suggested that lactate may serve as a metabolic regulator or intercellular signaling molecule rather than a fuel, modulating brain glucose metabolism, oxygen delivery and CBF [176, 177]. Mechanisms by which lactate might exert this effect [178] include modulation of prostaglandin action (and thus CBF) [177, 179], adjustment of the NADH/NAD+ redox ratio [123], and the regulation of neuronal cAMP formation via the lactate receptor G-protein-coupled receptor 81 (GPR81) [180].
Conclusion
Hypoglycemia is the principal barrier for achieving optimal, let alone normal, glycemic control for indefinite periods of time in patients with type 1 diabetes and advanced insulin-requiring type 2 diabetes [181]. Recurrent hypoglycemia forms the basis of HAAF and the clinical syndrome of impaired awareness of hypoglycemia by attenuating physiological defenses against subsequent hypoglycemia, consequently increasing the risk for severe hypoglycemia. Paradoxically, the mechanism(s) underlying these glucose counterregulatory impairments may be related to, or even caused by, processes that are seemingly aimed at protecting the brain against harm from severe hypoglycemia. The progress in metabolic and functional neuroimaging techniques has revealed that recurrent hypoglycemia causes cerebral adaptations to occur on many different levels. These adaptations include those in the regional delivery (blood flow) and transport of glucose to the brain, the handling of glucose by the brain and that of non-glucose alternative fuels, as well as activation or de-activation of brain areas involved in behavioral responses. It remains to be elucidated whether, and if so under which circumstances and in which brain areas, the brain uses non-glucose alternative sources of energy, particularly lactate, and whether this contributes to the emergence of impaired awareness of hypoglycemia. Such information is needed first to foster personalized decision-making with respect to glycemic targets, but should eventually lead to treatments that eliminate hypoglycemia from the lives of people with type 1 diabetes without compromising glucose control.
Abbreviations
- ASL:
-
Arterial spin labeling
- ANLS:
-
Astrocyte-neuron lactate shuttle
- BOLD:
-
Blood oxygenation level dependent
- CBF:
-
Cerebral blood flow
- CMRglc :
-
Cerebral metabolic rate of glucose
- CMRO2 :
-
Cerebral oxygen metabolic rate
- ECF:
-
Extracellular fluid
- FDG:
-
18F-Fluoro-2-deoxy-d-glucose
- fMRI:
-
Functional magnetic resonance imaging
- GABA:
-
Gamma-aminobutyric acid
- Glc-6-P:
-
Glucose-6-phosphate
- GLUT:
-
Glucose transporter protein
- GPR81:
-
G-protein-coupled receptor 81
- HAAF:
-
Hypoglycemia-associated autonomic failure
- HbA1c :
-
Glycosylated hemoglobin
- MCA:
-
Monocarboxylic acid
- MCTs:
-
Monocarboxylic acid transporters
- MRS:
-
Magnetic resonance spectroscopy
- NALS:
-
Neuron-astrocyte lactate shuttle
- PET:
-
Positron emission tomography
- RF:
-
Radiofrequency
- SNR:
-
Signal to noise ratio
- TCA:
-
Tricarboxylic acid
- VMH:
-
Ventromedial hypothalamus
- 3-OMG:
-
11C-3-O-methyl-d-glucose
- 13C:
-
Carbon-13
- 31P:
-
Phosphorus-31
References
Clarke DD, Sokoloff L (1999) Circulation and energy metabolism of the brain. In: Siegel GJ, Agrafnoff BW, Albers RW, Molinoff SK, Fisher PB, Uhler MD (eds) Basic neurochemistry: molecular, cellular and medical aspects, 6th edn. Lippincott-Raven, Philadelphia
(1993) The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med 329(14):977–986. doi:10.1056/NEJM199309303291401
Pedersen-Bjergaard U, Agerholm-Larsen B, Pramming S, Hougaard P, Thorsteinsson B (2001) Activity of angiotensin-converting enzyme and risk of severe hypoglycaemia in type 1 diabetes mellitus. Lancet 357(9264):1248–1253. doi:10.1016/S0140-6736(00)04405-6
Pedersen-Bjergaard U, Pramming S, Heller SR, Wallace TM, Rasmussen AK, Jorgensen HV, Matthews DR, Hougaard P, Thorsteinsson B (2004) Severe hypoglycaemia in 1076 adult patients with type 1 diabetes: influence of risk markers and selection. Diabetes Metab Res Rev 20(6):479–486. doi:10.1002/dmrr.482
Ostenson CG, Geelhoed-Duijvestijn P, Lahtela J, Weitgasser R, Markert Jensen M, Pedersen-Bjergaard U (2014) Self-reported non-severe hypoglycaemic events in Europe. Diabet Med 31(1):92–101. doi:10.1111/dme.12261
ter Braak EW, Appelman AM, van de Laak M, Stolk RP, van Haeften TW, Erkelens DW (2000) Clinical characteristics of type 1 diabetic patients with and without severe hypoglycemia. Diabetes Care 23(10):1467–1471
Group UKHS (2007) Risk of hypoglycaemia in types 1 and 2 diabetes: effects of treatment modalities and their duration. Diabetologia 50(6):1140–1147. doi:10.1007/s00125-007-0599-y
Gonder-Frederick LA, Cox DJ, Driesen NR, Ryan CM, Clarke WL (1994) Individual differences in neurobehavioral disruption during mild and moderate hypoglycemia in adults with IDDM. Diabetes 43(12):1407–1412
Cryer PE (2013) Mechanisms of hypoglycemia-associated autonomic failure in diabetes. N Engl J Med 369(4):362–372. doi:10.1056/NEJMra1215228
Cryer PE (2008) The barrier of hypoglycemia in diabetes. Diabetes 57(12):3169–3176. doi:10.2337/db08-1084
Tesfaye N, Seaquist ER (2010) Neuroendocrine responses to hypoglycemia. Ann N Y Acad Sci 1212:12–28. doi:10.1111/j.1749-6632.2010.05820.x
Gerich JE, Langlois M, Noacco C, Karam JH, Forsham PH (1973) Lack of glucagon response to hypoglycemia in diabetes: evidence for an intrinsic pancreatic alpha cell defect. Science 182(4108):171–173
Amiel SA, Sherwin RS, Simonson DC, Tamborlane WV (1988) Effect of intensive insulin therapy on glycemic thresholds for counterregulatory hormone release. Diabetes 37(7):901–907
Fritsche A, Stefan N, Haring H, Gerich J, Stumvoll M (2001) Avoidance of hypoglycemia restores hypoglycemia awareness by increasing beta-adrenergic sensitivity in type 1 diabetes. Ann Intern Med 134(9 Pt 1):729–736
Dagogo-Jack S, Rattarasarn C, Cryer PE (1994) Reversal of hypoglycemia unawareness, but not defective glucose counterregulation, in IDDM. Diabetes 43(12):1426–1434
Seaquist ER, Anderson J, Childs B, Cryer P, Dagogo-Jack S, Fish L, Heller SR, Rodriguez H, Rosenzweig J, Vigersky R (2013) Hypoglycemia and diabetes: a report of a workgroup of the American Diabetes Association and the Endocrine Society. Diabetes Care 36(5):1384–1395. doi:10.2337/dc12-2480
Geddes J, Schopman JE, Zammitt NN, Frier BM (2008) Prevalence of impaired awareness of hypoglycaemia in adults with Type 1 diabetes. Diabet Med 25(4):501–504. doi:10.1111/j.1464-5491.2008.02413.x
Schopman JE, Geddes J, Frier BM (2010) Prevalence of impaired awareness of hypoglycaemia and frequency of hypoglycaemia in insulin-treated type 2 diabetes. Diabetes Res Clin Pract 87(1):64–68. doi:10.1016/j.diabres.2009.10.013
Dagogo-Jack SE, Craft S, Cryer PE (1993) Hypoglycemia-associated autonomic failure in insulin-dependent diabetes mellitus. Recent antecedent hypoglycemia reduces autonomic responses to, symptoms of, and defense against subsequent hypoglycemia. J Clin Invest 91(3):819–828. doi:10.1172/JCI116302
Cranston I, Lomas J, Maran A, Macdonald I, Amiel SA (1994) Restoration of hypoglycaemia awareness in patients with long-duration insulin-dependent diabetes. Lancet 344(8918):283–287
Fanelli CG, Epifano L, Rambotti AM, Pampanelli S, Di Vincenzo A, Modarelli F, Lepore M, Annibale B, Ciofetta M, Bottini P et al (1993) Meticulous prevention of hypoglycemia normalizes the glycemic thresholds and magnitude of most of neuroendocrine responses to, symptoms of, and cognitive function during hypoglycemia in intensively treated patients with short-term IDDM. Diabetes 42(11):1683–1689
Zammitt NN, Warren RE, Deary IJ, Frier BM (2008) Delayed recovery of cognitive function following hypoglycemia in adults with type 1 diabetes: effect of impaired awareness of hypoglycemia. Diabetes 57(3):732–736. doi:10.2337/db07-0695
Graveling AJ, Deary IJ, Frier BM (2013) Acute hypoglycemia impairs executive cognitive function in adults with and without type 1 diabetes. Diabetes Care 36(10):3240–3246. doi:10.2337/dc13-0194
Inkster B, Frier BM (2012) The effects of acute hypoglycaemia on cognitive function in type 1 diabetes. Br J Diabetes Vasc Dis 12(5):221–226
Tkacs NC, Dunn-Meynell AA, Levin BE (2000) Presumed apoptosis and reduced arcuate nucleus neuropeptide Y and pro-opiomelanocortin mRNA in non-coma hypoglycemia. Diabetes 49(5):820–826
Ennis K, Tran PV, Seaquist ER, Rao R (2008) Postnatal age influences hypoglycemia-induced neuronal injury in the rat brain. Brain Res 1224:119–126. doi:10.1016/j.brainres.2008.06.003
Auer RN, Wieloch T, Olsson Y, Siesjo BK (1984) The distribution of hypoglycemic brain damage. Acta Neuropathol 64(3):177–191
Ma JH, Kim YJ, Yoo WJ, Ihn YK, Kim JY, Song HH, Kim BS (2009) MR imaging of hypoglycemic encephalopathy: lesion distribution and prognosis prediction by diffusion-weighted imaging. Neuroradiology 51(10):641–649. doi:10.1007/s00234-009-0544-5
Kang EG, Jeon SJ, Choi SS, Song CJ, Yu IK (2010) Diffusion MR imaging of hypoglycemic encephalopathy. AJNR Am J Neuroradiol 31(3):559–564. doi:10.3174/ajnr.A1856
Yoneda Y, Yamamoto S (2005) Cerebral cortical laminar necrosis on diffusion-weighted MRI in hypoglycaemic encephalopathy. Diabet Med 22(8):1098–1100. doi:10.1111/j.1464-5491.2005.01568.x
Jacobson AM, Musen G, Ryan CM, Silvers N, Cleary P, Waberski B, Burwood A, Weinger K, Bayless M, Dahms W, Harth J (2007) Long-term effect of diabetes and its treatment on cognitive function. N Engl J Med 356(18):1842–1852. doi:10.1056/NEJMoa066397
Nathan DM, Group DER (2014) The diabetes control and complications trial/epidemiology of diabetes interventions and complications study at 30 years: overview. Diabetes Care 37(1):9–16. doi:10.2337/dc13-2112
Skrivarhaug T, Bangstad HJ, Stene LC, Sandvik L, Hanssen KF, Joner G (2006) Long-term mortality in a nationwide cohort of childhood-onset type 1 diabetic patients in Norway. Diabetologia 49(2):298–305. doi:10.1007/s00125-005-0082-6
Patterson CC, Dahlquist G, Harjutsalo V, Joner G, Feltbower RG, Svensson J, Schober E, Gyurus E, Castell C, Urbonaite B, Rosenbauer J, Iotova V, Thorsson AV, Soltesz G (2007) Early mortality in EURODIAB population-based cohorts of type 1 diabetes diagnosed in childhood since 1989. Diabetologia 50(12):2439–2442. doi:10.1007/s00125-007-0824-8
Perantie DC, Lim A, Wu J, Weaver P, Warren SL, Sadler M, White NH, Hershey T (2008) Effects of prior hypoglycemia and hyperglycemia on cognition in children with type 1 diabetes mellitus. Pediatr Diabetes 9(2):87–95. doi:10.1111/j.1399-5448.2007.00274.x
Lin A, Northam EA, Rankins D, Werther GA, Cameron FJ (2010) Neuropsychological profiles of young people with type 1 diabetes 12 yr after disease onset. Pediatr Diabetes 11(4):235–243. doi:10.1111/j.1399-5448.2009.00588.x
Whitmer RA, Karter AJ, Yaffe K, Quesenberry CP Jr, Selby JV (2009) Hypoglycemic episodes and risk of dementia in older patients with type 2 diabetes mellitus. JAMA 301(15):1565–1572. doi:10.1001/jama.2009.460
Yaffe K, Falvey CM, Hamilton N, Harris TB, Simonsick EM, Strotmeyer ES, Shorr RI, Metti A, Schwartz AV, Health ABCS (2013) Association between hypoglycemia and dementia in a biracial cohort of older adults with diabetes mellitus. JAMA Intern Med 173(14):1300–1306. doi:10.1001/jamainternmed.2013.6176
Zoungas S, Patel A, Chalmers J, de Galan BE, Li Q, Billot L, Woodward M, Ninomiya T, Neal B, MacMahon S, Grobbee DE, Kengne AP, Marre M, Heller S, ADVANCE Collaborative Group (2010) Severe hypoglycemia and risks of vascular events and death. N Engl J Med 363(15):1410–1418. doi:10.1056/NEJMoa1003795
Goto A, Arah OA, Goto M, Terauchi Y, Noda M (2013) Severe hypoglycaemia and cardiovascular disease: systematic review and meta-analysis with bias analysis. BMJ 347:f4533. doi:10.1136/bmj.f4533
Mergenthaler P, Kahl A, Kamitz A, van Laak V, Stohlmann K, Thomsen S, Klawitter H, Przesdzing I, Neeb L, Freyer D, Priller J, Collins TJ, Megow D, Dirnagl U, Andrews DW, Meisel A (2012) Mitochondrial hexokinase II (HKII) and phosphoprotein enriched in astrocytes (PEA15) form a molecular switch governing cellular fate depending on the metabolic state. Proc Natl Acad Sci USA 109(5):1518–1523. doi:10.1073/pnas.1108225109
Wright RJ, Frier BM (2008) Vascular disease and diabetes: is hypoglycaemia an aggravating factor? Diabetes Metab Res Rev 24(5):353–363. doi:10.1002/dmrr.865
Rana O, Byrne CD, Kerr D, Coppini DV, Zouwail S, Senior R, Begley J, Walker JJ, Greaves K (2011) Acute hypoglycemia decreases myocardial blood flow reserve in patients with type 1 diabetes mellitus and in healthy humans. Circulation 124(14):1548–1556. doi:10.1161/CIRCULATIONAHA.110.992297
Gruden G, Barutta F, Chaturvedi N, Schalkwijk C, Stehouwer CD, Witte DR, Fuller JH, Perin PC, Bruno G (2012) Severe hypoglycemia and cardiovascular disease incidence in type 1 diabetes: the EURODIAB Prospective Complications Study. Diabetes Care 35(7):1598–1604. doi:10.2337/dc11-1531
Puente EC, Silverstein J, Bree AJ, Musikantow DR, Wozniak DF, Maloney S, Daphna-Iken D, Fisher SJ (2010) Recurrent moderate hypoglycemia ameliorates brain damage and cognitive dysfunction induced by severe hypoglycemia. Diabetes 59(4):1055–1062. doi:10.2337/db09-1495
Reno CM, Daphna-Iken D, Chen YS, VanderWeele J, Jethi K, Fisher SJ (2013) Severe hypoglycemia-induced lethal cardiac arrhythmias are mediated by sympathoadrenal activation. Diabetes 62(10):3570–3581. doi:10.2337/db13-0216
Sejling AS, Schouwenberg B, Faerch LH, Thorsteinsson B, de Galan BE, Pedersen-Bjergaard U (2015) Association between hypoglycaemia and impaired hypoglycaemia awareness and mortality in people with Type 1 diabetes mellitus. Diabet Med. doi:10.1111/dme.12810
Donovan CM, Watts AG (2014) Peripheral and central glucose sensing in hypoglycemic detection. Physiology (Bethesda) 29(5):314–324. doi:10.1152/physiol.00069.2013
Watts AG, Donovan CM (2010) Sweet talk in the brain: glucosensing, neural networks, and hypoglycemic counterregulation. Front Neuroendocrinol 31(1):32–43. doi:10.1016/j.yfrne.2009.10.006
Borg WP, During MJ, Sherwin RS, Borg MA, Brines ML, Shulman GI (1994) Ventromedial hypothalamic lesions in rats suppress counterregulatory responses to hypoglycemia. J Clin Invest 93(4):1677–1682. doi:10.1172/JCI117150
Borg MA, Sherwin RS, Borg WP, Tamborlane WV, Shulman GI (1997) Local ventromedial hypothalamus glucose perfusion blocks counterregulation during systemic hypoglycemia in awake rats. J Clin Invest 99(2):361–365. doi:10.1172/JCI119165
Borg WP, Sherwin RS, During MJ, Borg MA, Shulman GI (1995) Local ventromedial hypothalamus glucopenia triggers counterregulatory hormone release. Diabetes 44(2):180–184
Fery F, Plat L, van de Borne P, Cogan E, Mockel J (1999) Impaired counterregulation of glucose in a patient with hypothalamic sarcoidosis. N Engl J Med 340(11):852–856. doi:10.1056/NEJM199903183401105
McCrimmon R (2009) Glucose sensing during hypoglycemia: lessons from the lab. Diabetes Care 32(8):1357–1363. doi:10.2337/dc09-0123
Borg MA, Tamborlane WV, Shulman GI, Sherwin RS (2003) Local lactate perfusion of the ventromedial hypothalamus suppresses hypoglycemic counterregulation. Diabetes 52(3):663–666
Chan O, Paranjape SA, Horblitt A, Zhu W, Sherwin RS (2013) Lactate-induced release of GABA in the ventromedial hypothalamus contributes to counterregulatory failure in recurrent hypoglycemia and diabetes. Diabetes 62(12):4239–4246. doi:10.2337/db13-0770
Chan O, Sherwin R (2013) Influence of VMH fuel sensing on hypoglycemic responses. Trends Endocrinol Metab 24(12):616–624. doi:10.1016/j.tem.2013.08.005
Dienel GA (2012) Fueling and imaging brain activation. ASN Neuro 4(5). doi:10.1042/AN20120021
Simpson IA, Carruthers A, Vannucci SJ (2007) Supply and demand in cerebral energy metabolism: the role of nutrient transporters. J Cereb Blood Flow Metab 27(11):1766–1791. doi:10.1038/sj.jcbfm.9600521
Wilson JE (2003) Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol 206(Pt 12):2049–2057
Kety SS, Schmidt CF (1945) The determination of cerebral blood flow in man by the use of nitrous oxide in low concentrations. Am J Phsyiol 143:53–65
Kety SS, Schmidt CF (1948) The nitrous oxide method for the quantitative determination of cerebral blood flow in man: theory, procedure and normal values. J Clin Invest 27(4):476–483. doi:10.1172/JCI101994
Lassen NA (1985) Normal average value of cerebral blood flow in younger adults is 50 ml/100 g/min. J Cereb Blood Flow Metab 5(3):347–349. doi:10.1038/jcbfm.1985.48
Madsen PL, Holm S, Herning M, Lassen NA (1993) Average blood flow and oxygen uptake in the human brain during resting wakefulness: a critical appraisal of the Kety–Schmidt technique. J Cereb Blood Flow Metab 13(4):646–655. doi:10.1038/jcbfm.1993.83
Boyle PJ, Nagy RJ, O’Connor AM, Kempers SF, Yeo RA, Qualls C (1994) Adaptation in brain glucose uptake following recurrent hypoglycemia. Proc Natl Acad Sci USA 91(20):9352–9356
Boyle PJ, Kempers SF, O’Connor AM, Nagy RJ (1995) Brain glucose uptake and unawareness of hypoglycemia in patients with insulin-dependent diabetes mellitus. N Engl J Med 333(26):1726–1731. doi:10.1056/NEJM199512283332602
Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA (2010) Glial and neuronal control of brain blood flow. Nature 468(7321):232–243. doi:10.1038/nature09613
Zimmer L, Luxen A (2012) PET radiotracers for molecular imaging in the brain: past, present and future. Neuroimage 61(2):363–370. doi:10.1016/j.neuroimage.2011.12.037
Herscovitch P, Markham J, Raichle ME (1983) Brain blood flow measured with intravenous H2(15)O. I. Theory and error analysis. J Nucl Med 24(9):782–789
Baron JC, Jones T (2012) Oxygen metabolism, oxygen extraction and positron emission tomography: historical perspective and impact on basic and clinical neuroscience. Neuroimage 61(2):492–504. doi:10.1016/j.neuroimage.2011.12.036
Teves D, Videen TO, Cryer PE, Powers WJ (2004) Activation of human medial prefrontal cortex during autonomic responses to hypoglycemia. Proc Natl Acad Sci USA 101(16):6217–6221. doi:10.1073/pnas.0307048101
Bingham EM, Dunn JT, Smith D, Sutcliffe-Goulden J, Reed LJ, Marsden PK, Amiel SA (2005) Differential changes in brain glucose metabolism during hypoglycaemia accompany loss of hypoglycaemia awareness in men with type 1 diabetes mellitus. An [11C]-3-O-methyl-d-glucose PET study. Diabetologia 48(10):2080–2089. doi:10.1007/s00125-005-1900-6
Dunn JT, Cranston I, Marsden PK, Amiel SA, Reed LJ (2007) Attenuation of amydgala and frontal cortical responses to low blood glucose concentration in asymptomatic hypoglycemia in type 1 diabetes: a new player in hypoglycemia unawareness? Diabetes 56(11):2766–2773. doi:10.2337/db07-0666
Teh MM, Dunn JT, Choudhary P, Samarasinghe Y, Macdonald I, O’Doherty M, Marsden P, Reed LJ, Amiel SA (2010) Evolution and resolution of human brain perfusion responses to the stress of induced hypoglycemia. Neuroimage 53(2):584–592. doi:10.1016/j.neuroimage.2010.06.033
Reivich M, Kuhl D, Wolf A, Greenberg J, Phelps M, Ido T, Casella V, Fowler J, Hoffman E, Alavi A, Som P, Sokoloff L (1979) The [18F]fluorodeoxyglucose method for the measurement of local cerebral glucose utilization in man. Circ Res 44(1):127–137
Reivich M, Alavi A, Wolf A, Fowler J, Russell J, Arnett C, MacGregor RR, Shiue CY, Atkins H, Anand A et al (1985) Glucose metabolic rate kinetic model parameter determination in humans: the lumped constants and rate constants for [18F]fluorodeoxyglucose and [11C]deoxyglucose. J Cereb Blood Flow Metab 5(2):179–192. doi:10.1038/jcbfm.1985.24
Holden JE, Mori K, Dienel GA, Cruz NF, Nelson T, Sokoloff L (1991) Modeling the dependence of hexose distribution volumes in brain on plasma glucose concentration: implications for estimation of the local 2-deoxyglucose lumped constant. J Cereb Blood Flow Metab 11(2):171–182. doi:10.1038/jcbfm.1991.50
Suda S, Shinohara M, Miyaoka M, Lucignani G, Kennedy C, Sokoloff L (1990) The lumped constant of the deoxyglucose method in hypoglycemia: effects of moderate hypoglycemia on local cerebral glucose utilization in the rat. J Cereb Blood Flow Metab 10(4):499–509. doi:10.1038/jcbfm.1990.92
Nakanishi H, Cruz NF, Adachi K, Sokoloff L, Dienel GA (1996) Influence of glucose supply and demand on determination of brain glucose content with labeled methylglucose. J Cereb Blood Flow Metab 16(3):439–449. doi:10.1097/00004647-199605000-00010
Spring-Robinson C, Chandramouli V, Schumann WC, Faulhaber PF, Wang Y, Wu C, Ismail-Beigi F, Muzic RF Jr (2009) Uptake of 18F-labeled 6-fluoro-6-deoxy-d-glucose by skeletal muscle is responsive to insulin stimulation. J Nucl Med 50(6):912–919. doi:10.2967/jnumed.109.062687
Detre JA, Wang J (2002) Technical aspects and utility of fMRI using BOLD and ASL. Clin Neurophysiol 113(5):621–634
McKenna MC, Dienel GA, Sonnewald U, Waagepetersen HS, Schousboe A (2012) Energy metabolism of the brain. In: Wayne Albers R, Siegel GJ (eds) Basic neurochemistry, principles of molecular, cellular, and medical neurobiology, 8th edn. Elsevier, Amsterdam
Bolo NR, Musen G, Jacobson AM, Weinger K, McCartney RL, Flores V, Renshaw PF, Simonson DC (2011) Brain activation during working memory is altered in patients with type 1 diabetes during hypoglycemia. Diabetes 60(12):3256–3264. doi:10.2337/db11-0506
Rosenthal JM, Amiel SA, Yaguez L, Bullmore E, Hopkins D, Evans M, Pernet A, Reid H, Giampietro V, Andrew CM, Suckling J, Simmons A, Williams SC (2001) The effect of acute hypoglycemia on brain function and activation: a functional magnetic resonance imaging study. Diabetes 50(7):1618–1626
Anderson AW, Heptulla RA, Driesen N, Flanagan D, Goldberg PA, Jones TW, Rife F, Sarofin H, Tamborlane W, Sherwin R, Gore JC (2006) Effects of hypoglycemia on human brain activation measured with fMRI. Magn Reson Imaging 24(6):693–697. doi:10.1016/j.mri.2006.03.013
Detre JA, Wang J, Wang Z, Rao H (2009) Arterial spin-labeled perfusion MRI in basic and clinical neuroscience. Curr Opin Neurol 22(4):348–355. doi:10.1097/WCO.0b013e32832d9505
Alsop DC, Detre JA, Golay X, Gunther M, Hendrikse J, Hernandez-Garcia L, Lu H, Macintosh BJ, Parkes LM, Smits M, van Osch MJ, Wang DJ, Wong EC, Zaharchuk G (2014) Recommended implementation of arterial spin-labeled perfusion MRI for clinical applications: a consensus of the ISMRM perfusion study group and the European consortium for ASL in dementia. Magn Reson Med. doi:10.1002/mrm.25197
Boscolo Galazzo I, Storti SF, Formaggio E, Pizzini FB, Fiaschi A, Beltramello A, Bertoldo A, Manganotti P (2014) Investigation of brain hemodynamic changes induced by active and passive movements: a combined arterial spin labeling-BOLD fMRI study. J Magn Reson Imaging 40(4):937–948. doi:10.1002/jmri.24432
Di Costanzo A, Trojsi F, Tosetti M, Schirmer T, Lechner SM, Popolizio T, Scarabino T (2007) Proton MR spectroscopy of the brain at 3 T: an update. Eur Radiol 17(7):1651–1662. doi:10.1007/s00330-006-0546-1
van der Graaf M (2010) In vivo magnetic resonance spectroscopy: basic methodology and clinical applications. Eur Biophys J 39(4):527–540. doi:10.1007/s00249-009-0517-y
de Graaf RA, Mason GF, Patel AB, Behar KL, Rothman DL (2003) In vivo 1H-[13C]-NMR spectroscopy of cerebral metabolism. NMR Biomed 16(6–7):339–357. doi:10.1002/nbm.847
Hertz L, Gibbs ME, Dienel GA (2014) Fluxes of lactate into, from, and among gap junction-coupled astrocytes and their interaction with noradrenaline. Front Neurosci 8:261. doi:10.3389/fnins.2014.00261
Henry PG, Criego AB, Kumar A, Seaquist ER (2010) Measurement of cerebral oxidative glucose consumption in patients with type 1 diabetes mellitus and hypoglycemia unawareness using (13)C nuclear magnetic resonance spectroscopy. Metabolism 59(1):100–106. doi:10.1016/j.metabol.2009.07.012
van de Ven KC, de Galan BE, van der Graaf M, Shestov AA, Henry PG, Tack CJ, Heerschap A (2011) Effect of acute hypoglycemia on human cerebral glucose metabolism measured by (1)(3)C magnetic resonance spectroscopy. Diabetes 60(5):1467–1473. doi:10.2337/db10-1592
Lebon V, Petersen KF, Cline GW, Shen J, Mason GF, Dufour S, Behar KL, Shulman GI, Rothman DL (2002) Astroglial contribution to brain energy metabolism in humans revealed by 13C nuclear magnetic resonance spectroscopy: elucidation of the dominant pathway for neurotransmitter glutamate repletion and measurement of astrocytic oxidative metabolism. J Neurosci 22(5):1523–1531
Gulanski BI, De Feyter HM, Page KA, Belfort-DeAguiar R, Mason GF, Rothman DL, Sherwin RS (2013) Increased brain transport and metabolism of acetate in hypoglycemia unawareness. J Clin Endocrinol Metab 98(9):3811–3820. doi:10.1210/jc.2013-1701
Mason GF, Petersen KF, Lebon V, Rothman DL, Shulman GI (2006) Increased brain monocarboxylic acid transport and utilization in type 1 diabetes. Diabetes 55(4):929–934
de Graaf RA (2007) In vivo NMR spectroscopy: principles and techniques, 2nd edn. Wiley, West Sussex
Bischof MG, Mlynarik V, Brehm A, Bernroider E, Krssak M, Bauer E, Madl C, Bayerle-Eder M, Waldhausl W, Roden M (2004) Brain energy metabolism during hypoglycaemia in healthy and type 1 diabetic subjects. Diabetologia 47(4):648–651. doi:10.1007/s00125-004-1362-2
de Graaf RA, Pan JW, Telang F, Lee JH, Brown P, Novotny EJ, Hetherington HP, Rothman DL (2001) Differentiation of glucose transport in human brain gray and white matter. J Cereb Blood Flow Metab 21(5):483–492. doi:10.1097/00004647-200105000-00002
Gruetter R, Ugurbil K, Seaquist ER (1998) Steady-state cerebral glucose concentrations and transport in the human brain. J Neurochem 70(1):397–408
Choi IY, Lee SP, Kim SG, Gruetter R (2001) In vivo measurements of brain glucose transport using the reversible Michaelis–Menten model and simultaneous measurements of cerebral blood flow changes during hypoglycemia. J Cereb Blood Flow Metab 21(6):653–663. doi:10.1097/00004647-200106000-00003
Seaquist ER, Damberg GS, Tkac I, Gruetter R (2001) The effect of insulin on in vivo cerebral glucose concentrations and rates of glucose transport/metabolism in humans. Diabetes 50(10):2203–2209
van de Ven KC, van der Graaf M, Tack CJ, Heerschap A, de Galan BE (2012) Steady-state brain glucose concentrations during hypoglycemia in healthy humans and patients with type 1 diabetes. Diabetes 61(8):1974–1977. doi:10.2337/db11-1778
Lee DH, Chung MY, Lee JU, Kang DG, Paek YW (2000) Changes of glucose transporters in the cerebral adaptation to hypoglycemia. Diabetes Res Clin Pract 47(1):15–23
Uehara Y, Nipper V, McCall AL (1997) Chronic insulin hypoglycemia induces GLUT-3 protein in rat brain neurons. Am J Physiol 272(4 Pt 1):E716–E719
Simpson IA, Appel NM, Hokari M, Oki J, Holman GD, Maher F, Koehler-Stec EM, Vannucci SJ, Smith QR (1999) Blood–brain barrier glucose transporter: effects of hypo- and hyperglycemia revisited. J Neurochem 72(1):238–247
Kumagai AK, Kang YS, Boado RJ, Pardridge WM (1995) Upregulation of blood–brain barrier GLUT1 glucose transporter protein and mRNA in experimental chronic hypoglycemia. Diabetes 44(12):1399–1404
Alf MF, Duarte JM, Schibli R, Gruetter R, Kramer SD (2013) Brain glucose transport and phosphorylation under acute insulin-induced hypoglycemia in mice: an 18F-FDG PET study. J Nucl Med 54(12):2153–2160. doi:10.2967/jnumed.113.122812
Criego AB, Tkac I, Kumar A, Thomas W, Gruetter R, Seaquist ER (2005) Brain glucose concentrations in patients with type 1 diabetes and hypoglycemia unawareness. J Neurosci Res 79(1–2):42–47. doi:10.1002/jnr.20296
Criego AB, Tkac I, Kumar A, Thomas W, Gruetter R, Seaquist ER (2005) Brain glucose concentrations in healthy humans subjected to recurrent hypoglycemia. J Neurosci Res 82(4):525–530. doi:10.1002/jnr.20654
Segel SA, Fanelli CG, Dence CS, Markham J, Videen TO, Paramore DS, Powers WJ, Cryer PE (2001) Blood-to-brain glucose transport, cerebral glucose metabolism, and cerebral blood flow are not increased after hypoglycemia. Diabetes 50(8):1911–1917
Bouteldja N, Andersen LT, Moller N, Gormsen LC (2014) Using positron emission tomography to study human ketone body metabolism: a review. Metabolism 63(11):1375–1384. doi:10.1016/j.metabol.2014.08.001
Mitchell GA, Kassovska-Bratinova S, Boukaftane Y, Robert MF, Wang SP, Ashmarina L, Lambert M, Lapierre P, Potier E (1995) Medical aspects of ketone body metabolism. Clin Invest Med 18(3):193–216
Klepper J, Diefenbach S, Kohlschutter A, Voit T (2004) Effects of the ketogenic diet in the glucose transporter 1 deficiency syndrome. Prostaglandins Leukot Essent Fatty Acids 70(3):321–327. doi:10.1016/j.plefa.2003.07.004
Beylot M (1996) Regulation of in vivo ketogenesis: role of free fatty acids and control by epinephrine, thyroid hormones, insulin and glucagon. Diabetes Metab 22(5):299–304
Abi-Saab WM, Maggs DG, Jones T, Jacob R, Srihari V, Thompson J, Kerr D, Leone P, Krystal JH, Spencer DD, During MJ, Sherwin RS (2002) Striking differences in glucose and lactate levels between brain extracellular fluid and plasma in conscious human subjects: effects of hyperglycemia and hypoglycemia. J Cereb Blood Flow Metab 22(3):271–279. doi:10.1097/00004647-200203000-00004
Cooperberg BA, Cryer PE (2010) Insulin reciprocally regulates glucagon secretion in humans. Diabetes 59(11):2936–2940. doi:10.2337/db10-0728
Boumezbeur F, Petersen KF, Cline GW, Mason GF, Behar KL, Shulman GI, Rothman DL (2010) The contribution of blood lactate to brain energy metabolism in humans measured by dynamic 13C nuclear magnetic resonance spectroscopy. J Neurosci 30(42):13983–13991. doi:10.1523/JNEUROSCI.2040-10.2010
van Hall G, Stromstad M, Rasmussen P, Jans O, Zaar M, Gam C, Quistorff B, Secher NH, Nielsen HB (2009) Blood lactate is an important energy source for the human brain. J Cereb Blood Flow Metab 29(6):1121–1129. doi:10.1038/jcbfm.2009.35
Patil GD, Briski KP (2005) Lactate is a critical “sensed” variable in caudal hindbrain monitoring of CNS metabolic stasis. Am J Physiol Regul Integr Comp Physiol 289(6):R1777–R1786. doi:10.1152/ajpregu.00177.2004
Pellerin L, Magistretti PJ (1994) Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci USA 91(22):10625–10629
Brooks GA (2009) Cell–cell and intracellular lactate shuttles. J Physiol 587(Pt 23):5591–5600. doi:10.1113/jphysiol.2009.178350
Jolivet R, Allaman I, Pellerin L, Magistretti PJ, Weber B (2010) Comment on recent modeling studies of astrocyte-neuron metabolic interactions. J Cereb Blood Flow Metab 30(12):1982–1986. doi:10.1038/jcbfm.2010.132
Mangia S, DiNuzzo M, Giove F, Carruthers A, Simpson IA, Vannucci SJ (2011) Response to ‘comment on recent modeling studies of astrocyte-neuron metabolic interactions’: much ado about nothing. J Cereb Blood Flow Metab 31(6):1346–1353. doi:10.1038/jcbfm.2011.29
Lundgaard I, Li B, Xie L, Kang H, Sanggaard S, Haswell JD, Sun W, Goldman S, Blekot S, Nielsen M, Takano T, Deane R, Nedergaard M (2015) Direct neuronal glucose uptake heralds activity-dependent increases in cerebral metabolism. Nat Commun 6:6807. doi:10.1038/ncomms7807
Duarte JM, Girault FM, Gruetter R (2015) Brain energy metabolism measured by (13) C magnetic resonance spectroscopy in vivo upon infusion of [3-(13) C]lactate. J Neurosci Res 93(7):1009–1018. doi:10.1002/jnr.23531
Aveseh M, Nikooie R, Sheibani V, Mahani SE (2014) Endurance training increases brain lactate uptake during hypoglycemia by up regulation of brain lactate transporters. Mol Cell Endocrinol. doi:10.1016/j.mce.2014.06.019
Herzog RI, Jiang L, Herman P, Zhao C, Sanganahalli BG, Mason GF, Hyder F, Rothman DL, Sherwin RS, Behar KL (2013) Lactate preserves neuronal metabolism and function following antecedent recurrent hypoglycemia. J Clin Invest 123(5):1988–1998. doi:10.1172/JCI65105
De Feyter HM, Mason GF, Shulman GI, Rothman DL, Petersen KF (2013) Increased brain lactate concentrations without increased lactate oxidation during hypoglycemia in type 1 diabetic individuals. Diabetes 62(9):3075–3080. doi:10.2337/db13-0313
Rossetti P, Porcellati F, Busciantella Ricci N, Candeloro P, Cioli P, Nair KS, Santeusanio F, Bolli GB, Fanelli CG (2008) Effect of oral amino acids on counterregulatory responses and cognitive function during insulin-induced hypoglycemia in nondiabetic and type 1 diabetic people. Diabetes 57(7):1905–1917. doi:10.2337/db08-0276
Evans ML, Hopkins D, Macdonald IA, Amiel SA (2004) Alanine infusion during hypoglycaemia partly supports cognitive performance in healthy human subjects. Diabet Med 21(5):440–446. doi:10.1111/j.1464-5491.2004.01174.x
Schmoll D, Fuhrmann E, Gebhardt R, Hamprecht B (1995) Significant amounts of glycogen are synthesized from 3-carbon compounds in astroglial primary cultures from mice with participation of the mitochondrial phosphoenolpyruvate carboxykinase isoenzyme. Eur J Biochem 227(1–2):308–315
Wong KL, Tyce GM (1983) Glucose and amino acid metabolism in rat brain during sustained hypoglycemia. Neurochem Res 8(4):401–415
Wahren J, Ekberg K, Fernqvist-Forbes E, Nair S (1999) Brain substrate utilisation during acute hypoglycaemia. Diabetologia 42(7):812–818
Lubow JM, Pinon IG, Avogaro A, Cobelli C, Treeson DM, Mandeville KA, Toffolo G, Boyle PJ (2006) Brain oxygen utilization is unchanged by hypoglycemia in normal humans: lactate, alanine, and leucine uptake are not sufficient to offset energy deficit. Am J Physiol Endocrinol Metab 290(1):E149–E153. doi:10.1152/ajpendo.00049.2005
Ebert D, Haller RG, Walton ME (2003) Energy contribution of octanoate to intact rat brain metabolism measured by 13C nuclear magnetic resonance spectroscopy. J Neurosci 23(13):5928–5935
Evans ML, Matyka K, Lomas J, Pernet A, Cranston IC, Macdonald I, Amiel SA (1998) Reduced counterregulation during hypoglycemia with raised circulating nonglucose lipid substrates: evidence for regional differences in metabolic capacity in the human brain? J Clin Endocrinol Metab 83(8):2952–2959. doi:10.1210/jcem.83.8.4937
Page KA, Williamson A, Yu N, McNay EC, Dzuira J, McCrimmon RJ, Sherwin RS (2009) Medium-chain fatty acids improve cognitive function in intensively treated type 1 diabetic patients and support in vitro synaptic transmission during acute hypoglycemia. Diabetes 58(5):1237–1244. doi:10.2337/db08-1557
Cheah YS, Amiel SA (2012) Metabolic neuroimaging of the brain in diabetes mellitus and hypoglycaemia. Nat Rev Endocrinol 8(10):588–597. doi:10.1038/nrendo.2012.97
Cranston I, Reed LJ, Marsden PK, Amiel SA (2001) Changes in regional brain (18)F-fluorodeoxyglucose uptake at hypoglycemia in type 1 diabetic men associated with hypoglycemia unawareness and counter-regulatory failure. Diabetes 50(10):2329–2336
Klomp DW, Renema WK, van der Graaf M, de Galan BE, Kentgens AP, Heerschap A (2006) Sensitivity-enhanced 13C MR spectroscopy of the human brain at 3 Tesla. Magn Reson Med 55(2):271–278. doi:10.1002/mrm.20745
van de Ven KC, van der Graaf M, Tack CJ, Klomp DW, Heerschap A, de Galan BE (2010) Optimized [1-(13)C]glucose infusion protocol for 13C magnetic resonance spectroscopy at 3T of human brain glucose metabolism under euglycemic and hypoglycemic conditions. J Neurosci Methods 186(1):68–71. doi:10.1016/j.jneumeth.2009.10.025
van de Ven KC, Tack CJ, Heerschap A, van der Graaf M, de Galan BE (2013) Patients with type 1 diabetes exhibit altered cerebral metabolism during hypoglycemia. J Clin Invest 123(2):623–629. doi:10.1172/JCI62742
Saez I, Duran J, Sinadinos C, Beltran A, Yanes O, Tevy MF, Martinez-Pons C, Milan M, Guinovart JJ (2014) Neurons have an active glycogen metabolism that contributes to tolerance to hypoxia. J Cereb Blood Flow Metab 34(6):945–955. doi:10.1038/jcbfm.2014.33
Choi IY, Seaquist ER, Gruetter R (2003) Effect of hypoglycemia on brain glycogen metabolism in vivo. J Neurosci Res 72(1):25–32. doi:10.1002/jnr.10574
Oz G, Kumar A, Rao JP, Kodl CT, Chow L, Eberly LE, Seaquist ER (2009) Human brain glycogen metabolism during and after hypoglycemia. Diabetes 58(9):1978–1985. doi:10.2337/db09-0226
Herzog RI, Chan O, Yu S, Dziura J, McNay EC, Sherwin RS (2008) Effect of acute and recurrent hypoglycemia on changes in brain glycogen concentration. Endocrinology 149(4):1499–1504. doi:10.1210/en.2007-1252
Oz G, Tesfaye N, Kumar A, Deelchand DK, Eberly LE, Seaquist ER (2012) Brain glycogen content and metabolism in subjects with type 1 diabetes and hypoglycemia unawareness. J Cereb Blood Flow Metab 32(2):256–263. doi:10.1038/jcbfm.2011.138
McKenna MC (2007) The glutamate-glutamine cycle is not stoichiometric: fates of glutamate in brain. J Neurosci Res 85(15):3347–3358. doi:10.1002/jnr.21444
Sonnewald U (2014) Glutamate synthesis has to be matched by its degradation—where do all the carbons go? J Neurochem 131(4):399–406. doi:10.1111/jnc.12812
Dienel GA, McKenna MC (2014) A dogma-breaking concept: glutamate oxidation in astrocytes is the source of lactate during aerobic glycolysis in resting subjects. J Neurochem. doi:10.1111/jnc.12835
Garcia-Espinosa MA, Rodrigues TB, Sierra A, Benito M, Fonseca C, Gray HL, Bartnik BL, Garcia-Martin ML, Ballesteros P, Cerdan S (2004) Cerebral glucose metabolism and the glutamine cycle as detected by in vivo and in vitro 13C NMR spectroscopy. Neurochem Int 45(2–3):297–303. doi:10.1016/j.neuint.2003.08.014
Bischof MG, Brehm A, Bernroider E, Krssak M, Mlynarik V, Krebs M, Roden M (2006) Cerebral glutamate metabolism during hypoglycaemia in healthy and type 1 diabetic humans. Eur J Clin Invest 36(3):164–169. doi:10.1111/j.1365-2362.2006.01615.x
Terpstra M, Moheet A, Kumar A, Eberly LE, Seaquist E, Oz G (2014) Changes in human brain glutamate concentration during hypoglycemia: insights into cerebral adaptations in hypoglycemia-associated autonomic failure in type 1 diabetes. J Cereb Blood Flow Metab 34(5):876–882. doi:10.1038/jcbfm.2014.32
Jiang L, Herzog RI, Mason GF, de Graaf RA, Rothman DL, Sherwin RS, Behar KL (2009) Recurrent antecedent hypoglycemia alters neuronal oxidative metabolism in vivo. Diabetes 58(6):1266–1274. doi:10.2337/db08-1664
Veneman T, Mitrakou A, Mokan M, Cryer P, Gerich J (1994) Effect of hyperketonemia and hyperlacticacidemia on symptoms, cognitive dysfunction, and counterregulatory hormone responses during hypoglycemia in normal humans. Diabetes 43(11):1311–1317
King P, Parkin H, Macdonald IA, Barber C, Tattersall RB (1997) The effect of intravenous lactate on cerebral function during hypoglycaemia. Diabet Med 14(1):19–28. doi:10.1002/(SICI)1096-9136(199701)14:1<19:AID-DIA289>3.0.CO;2-0
Maran A, Cranston I, Lomas J, Macdonald I, Amiel SA (1994) Protection by lactate of cerebral function during hypoglycaemia. Lancet 343(8888):16–20
Maran A, Crepaldi C, Trupiani S, Lucca T, Jori E, Macdonald IA, Tiengo A, Avogaro A, Del Prato S (2000) Brain function rescue effect of lactate following hypoglycaemia is not an adaptation process in both normal and type I diabetic subjects. Diabetologia 43(6):733–741. doi:10.1007/s001250051371
Tallroth G, Ryding E, Agardh CD (1992) Regional cerebral blood flow in normal man during insulin-induced hypoglycemia and in the recovery period following glucose infusion. Metabolism 41(7):717–721
Eckert B, Ryding E, Agardh CD (1998) Sustained elevation of cerebral blood flow after hypoglycaemia in normal man. Diabetes Res Clin Pract 40(2):91–100
Tallroth G, Ryding E, Agardh CD (1993) The influence of hypoglycaemia on regional cerebral blood flow and cerebral volume in type 1 (insulin-dependent) diabetes mellitus. Diabetologia 36(6):530–535
MacLeod KM, Gold AE, Ebmeier KP, Hepburn DA, Deary IJ, Goodwin GM, Frier BM (1996) The effects of acute hypoglycemia on relative cerebral blood flow distribution in patients with type I (insulin-dependent) diabetes and impaired hypoglycemia awareness. Metabolism 45(8):974–980
MacLeod KM, Hepburn DA, Deary IJ, Goodwin GM, Dougall N, Ebmeier KP, Frier BM (1994) Regional cerebral blood flow in IDDM patients: effects of diabetes and of recurrent severe hypoglycaemia. Diabetologia 37(3):257–263
Arbelaez AM, Su Y, Thomas JB, Hauch AC, Hershey T, Ances BM (2013) Comparison of regional cerebral blood flow responses to hypoglycemia using pulsed arterial spin labeling and positron emission tomography. PLoS One 8(3):e60085. doi:10.1371/journal.pone.0060085
Mangia S, Tesfaye N, De Martino F, Kumar AF, Kollasch P, Moheet AA, Eberly LE, Seaquist ER (2012) Hypoglycemia-induced increases in thalamic cerebral blood flow are blunted in subjects with type 1 diabetes and hypoglycemia unawareness. J Cereb Blood Flow Metab 32(11):2084–2090. doi:10.1038/jcbfm.2012.117
Musen G, Simonson DC, Bolo NR, Driscoll A, Weinger K, Raji A, Theberge J, Renshaw PF, Jacobson AM (2008) Regional brain activation during hypoglycemia in type 1 diabetes. J Clin Endocrinol Metab 93(4):1450–1457. doi:10.1210/jc.2007-2007
Page KA, Arora J, Qiu M, Relwani R, Constable RT, Sherwin RS (2009) Small decrements in systemic glucose provoke increases in hypothalamic blood flow prior to the release of counterregulatory hormones. Diabetes 58(2):448–452. doi:10.2337/db08-1224
Arbelaez AM, Powers WJ, Videen TO, Price JL, Cryer PE (2008) Attenuation of counterregulatory responses to recurrent hypoglycemia by active thalamic inhibition: a mechanism for hypoglycemia-associated autonomic failure. Diabetes 57(2):470–475. doi:10.2337/db07-1329
Newman EA (2013) Functional hyperemia and mechanisms of neurovascular coupling in the retinal vasculature. J Cereb Blood Flow Metab 33(11):1685–1695. doi:10.1038/jcbfm.2013.145
Vetri F, Xu H, Paisansathan C, Pelligrino DA (2012) Impairment of neurovascular coupling in type 1 diabetes mellitus in rats is linked to PKC modulation of BK(Ca) and Kir channels. Am J Physiol Heart Circ Physiol 302(6):H1274–H1284. doi:10.1152/ajpheart.01067.2011
Wessels AM, Rombouts SA, Simsek S, Kuijer JP, Kostense PJ, Barkhof F, Scheltens P, Snoek FJ, Heine RJ (2006) Microvascular disease in type 1 diabetes alters brain activation: a functional magnetic resonance imaging study. Diabetes 55(2):334–340
Larrabee MG (1995) Lactate metabolism and its effects on glucose metabolism in an excised neural tissue. J Neurochem 64(4):1734–1741
Wyss MT, Jolivet R, Buck A, Magistretti PJ, Weber B (2011) In vivo evidence for lactate as a neuronal energy source. J Neurosci 31(20):7477–7485. doi:10.1523/JNEUROSCI.0415-11.2011
Litvin M, Clark AL, Fisher SJ (2013) Recurrent hypoglycemia: boosting the brain’s metabolic flexibility. J Clin Invest 123(5):1922–1924. doi:10.1172/JCI69796
Vafaee MS, Vang K, Bergersen LH, Gjedde A (2012) Oxygen consumption and blood flow coupling in human motor cortex during intense finger tapping: implication for a role of lactate. J Cereb Blood Flow Metab 32(10):1859–1868. doi:10.1038/jcbfm.2012.89
Bergersen LH, Gjedde A (2012) Is lactate a volume transmitter of metabolic states of the brain? Front Neuroenergetics 4:5. doi:10.3389/fnene.2012.00005
Gordon GR, Choi HB, Rungta RL, Ellis-Davies GC, MacVicar BA (2008) Brain metabolism dictates the polarity of astrocyte control over arterioles. Nat New Biol 456(7223):745–749. doi:10.1038/nature07525
Lauritzen KH, Morland C, Puchades M, Holm-Hansen S, Hagelin EM, Lauritzen F, Attramadal H, Storm-Mathisen J, Gjedde A, Bergersen LH (2013) Lactate receptor sites link neurotransmission, neurovascular coupling, and brain energy metabolism. Cereb Cortex. doi:10.1093/cercor/bht136
Cryer PE (2008) Hypoglycemia: still the limiting factor in the glycemic management of diabetes. Endocr Pract 14(6):750–756. doi:10.4158/EP.14.6.750
Acknowledgments
We are indebted to Prof. A. Heerschap for his helpful advice. This work was financially supported by the Dutch Diabetes Research Foundation and the European Foundation for the Study of Diabetes (EFSD).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Rooijackers, H.M.M., Wiegers, E.C., Tack, C.J. et al. Brain glucose metabolism during hypoglycemia in type 1 diabetes: insights from functional and metabolic neuroimaging studies. Cell. Mol. Life Sci. 73, 705–722 (2016). https://doi.org/10.1007/s00018-015-2079-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-015-2079-8