
Abstract
Background
The pharmacokinetics and tolerability of semaglutide, a once-weekly human glucagon-like peptide-1 analog in development for the treatment of type 2 diabetes mellitus, were investigated in subjects with/without renal impairment (RI).
Methods
Fifty-six subjects, categorized into renal function groups [normal, mild, moderate, severe, and end-stage renal disease (ESRD)], received a single subcutaneous dose of semaglutide 0.5 mg. Semaglutide plasma concentrations were assessed ≤480 h post-dose; the primary endpoint was the area under the plasma concentration–time curve from time zero to infinity.
Results
Semaglutide exposure in subjects with mild/moderate RI and ESRD was similar to that in subjects with normal renal function. In subjects with severe RI, the mean exposure of semaglutide was 22% higher than in subjects with normal renal function, and the 95% confidence interval (1.02–1.47) for the ratio exceeded the pre-specified limits (0.70–1.43). When adjusted for differences in sex, age, and body weight between the groups, all comparisons were within the pre-specified clinically relevant limits. Across RI groups there was no relationship between creatinine clearance (CLCR) and semaglutide exposure, or between CLCR and semaglutide maximum plasma drug concentration (C max). Hemodialysis did not appear to affect the pharmacokinetics of semaglutide. No appreciable changes in safety parameters or vital signs and no serious adverse events were noted. One subject with severe RI reported two major hypoglycemic events.
Conclusion
When adjusted for differences in sex, age, and body weight, semaglutide exposure was similar between subjects with RI and subjects with normal renal function. Semaglutide (0.5 mg) was well-tolerated. Dose adjustment may not be warranted for subjects with RI.
ClinicalTrials.gov identifier
NCT00833716.
Similar content being viewed by others

Semaglutide exposure was similar between subjects with mild/moderate renal impairment (RI) or end-stage renal disease and subjects with normal renal function; equivalence was not demonstrated in subjects with severe RI, in whom mean exposure was 22% higher. However, when exposures were adjusted for differences in age, sex, and body weight, all comparisons were within the pre-specified ‘no effect’ limits. |
A single subcutaneous dose of semaglutide 0.5 mg was well-tolerated across all renal function groups. |
Semaglutide appears to be a useful treatment for subjects with diabetes mellitus regardless of renal status, and may not require dose adjustment. |
1 Introduction
Impaired renal function is a common complication of type 2 diabetes mellitus [1,2,3]. The prevalence of chronic renal disease, characterized by albuminuria or impaired renal function, is reported to be >30% among adults with type 2 diabetes, and duration of diabetes is associated with progression of renal disease [3,4,5,6,7,8]. Appropriate control of blood glucose is known to slow down the progress of renal impairment (RI) in patients with type 2 diabetes and kidney dysfunction [9, 10].
The metabolism and excretion of antidiabetic drugs required for good glycemic control may, however, be influenced by concomitant RI [10]. Reduced renal clearance, increased elimination half-life (t ½), and high maximum plasma drug concentration (C max) values of antidiabetic drugs may lead to high and prolonged drug exposure [11,12,13,14,15,16,17]. For some antidiabetic drugs, this increases the risk of hypoglycemia, meaning that they are unsuitable for use in patients with RI or that caution is advised [11,12,13,14,15,16,17]. Therapeutic options in this particular patient group are therefore limited.
Semaglutide (Novo Nordisk A/S, Denmark) is a human glucagon-like peptide-1 (GLP-1) analog in phase III clinical development for the treatment of type 2 diabetes [18]. Although native human GLP-1 increases insulin secretion and decreases glucagon secretion in a glucose-dependent manner, its short t ½ makes it unsuitable for clinical use in type 2 diabetes [19, 20]. Furthermore, as GLP-1 is cleared primarily via the kidney, it may be particularly unsuitable for use in patients with RI [19, 20]. While semaglutide has 94% structural homology to native human GLP-1, modifications have been made to extend its t ½ to approximately 1 week in humans, allowing once-weekly administration [18, 21]. These modifications include acylation of the peptide backbone with a spacer and a C-18 fatty di-acid chain to lysine at position 26, which mediates strong binding to albumin and potentially results in reduced renal clearance, and an amino-acid substitution in position 8, which results in reduced susceptibility to dipeptidyl peptidase-4 (DPP-4) degradation. Studies show limited renal excretion of semaglutide in its intact form [22]. Semaglutide monotherapy has been shown to significantly improve glycemic control and reduce body weight, with a low risk of hypoglycemia in subjects with type 2 diabetes [18, 21, 23].
Several studies have assessed the effect of RI on the pharmacokinetics of GLP-1 receptor agonists [11, 12, 24,25,26,27,28]. Pharmacokinetic, efficacy, and safety data indicate that liraglutide and albiglutide have favorable benefit–risk profiles in patients with type 2 diabetes who also have mild or moderate RI [24, 25, 29]. Both medications have been reported to show little or no change in pharmacokinetics and pharmacodynamics in patients with RI versus those with normal renal function, are well-tolerated, and exhibit a low risk of hypoglycemia, irrespective of renal function [24, 25, 29, 30]. Patients with concomitant type 2 diabetes and RI are expected to be able to use standard treatment regimens without the need for dose adjustments [24, 25, 30].
The aim of this study was to compare the pharmacokinetics of semaglutide in subjects with different degrees of RI versus those with normal renal function, to determine whether semaglutide dose adjustment may be required. The short-term safety and tolerability of semaglutide was also assessed.
2 Methods
2.1 Study Population
Study participants were of both sexes, 18–75 years of age, with a body mass index of ≤40 kg/m2. At screening, subjects had normal renal function or RI according to predefined creatinine clearance (CLCR) criteria using the Cockcroft–Gault formula [31]. Subjects were classified as one of five groups: normal renal function (CLCR > 80 mL/min); mild RI (CLCR > 50 and ≤80 mL/min), moderate RI (CLCR > 30 and ≤50 mL/min), severe RI (CLCR ≤ 30 mL/min), or subjects with end-stage renal disease (ESRD) requiring hemodialysis. Subjects with type 2 diabetes were allowed to participate in this study.
Renal transplant patients were excluded, as were those with serious cardiac disease [New York Heart Association (NYHA) heart failure functional class III or IV; myocardial infarction within 3 months; unstable angina pectoris], uncontrolled hypertension (diastolic blood pressure ≥100 mmHg or systolic blood pressure ≥180 mmHg), severe hepatic disease within the previous 12 months, and those with elevated liver enzymes (≥2.5 times upper normal range). Medications known to alter tubular secretion of creatinine were not permitted within either 14 days or five half-lives, whichever was longer, prior to dosing.
2.2 Treatment
Semaglutide was administered by subcutaneous injection in the thigh (NordiPen®, Novo Nordisk A/S, Denmark) as a single 0.5 mg dose. In the ESRD group, semaglutide was administered 1–24 h after hemodialysis, with no hemodialysis for 48 h post-dose.
Initially, a single dose of 10 µg/kg (absolute dose range 0.7–1.2 mg) was administered to six subjects (three with severe RI and three with ESRD) but, based on the frequency and severity of gastrointestinal adverse effects, the 10 µg/kg dose was not considered a tolerable starting dose. Two of these six subjects experienced three serious adverse events (AEs) (angioedema, increased hypertension, and aggravated sciatica pain), three experienced minor or symptoms-only hypoglycemia, and five experienced significant nausea and/or vomiting. To improve tolerability, the study was re-started with a lower, fixed single dose of semaglutide 0.5 mg.
2.3 Study Design and Pharmacokinetic Sampling
This was a multicenter, single-dose, open-label, parallel-group study (ClinicalTrials.gov identifier NCT00833716) conducted in accordance with Good Clinical Practice [32], the Declaration of Helsinki, and relevant guidelines for studies in patients with RI [33, 34]. A sample size of 54 subjects (14 normal renal function and ten in each of the four RI groups) was calculated to be sufficient for the primary objective of the study, with a power of 82% to show equivalence of exposure between subjects with severe RI and subjects with normal renal function. All participants provided written consent prior to initiation of any study-related activities. Participants were informed of the risks and benefits of the study and were permitted to withdraw at any time.
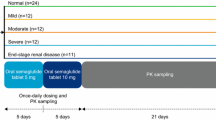
The study period was from Day 1 (dosing) to Day 21 (follow-up visit). Participants remained in-house for a minimum of 72 h after dosing (Fig. 1).

Study design. In the ESRD group, semaglutide was given 1–24 h after hemodialysis, with no hemodialysis for 48 h post-dose. ESRD end-stage renal disease, Max maximum, SC subcutaneous
To determine the plasma concentrations of semaglutide, blood samples were taken 15 min prior to administration of semaglutide and 2, 8, 14, 16, 18, 20, 22, 24, 26, 28, 30, 32, 34, 38, 42, 48, 54, 60, 66, 72, 96, 144, 192, 240, 360, and 480 h after administration of semaglutide. For ESRD subjects on hemodialysis, hourly blood samples (some of which were in addition to those mentioned above) were drawn during each of the first two hemodialysis sessions post-administration (each hemodialysis session lasted for ~2–4 h).
2.4 Assay Methodology
Semaglutide serum concentrations were measured by liquid chromatography with tandem mass spectrometry (Celerion Inc., Fehraltorf, Switzerland), as described elsewhere [21]. The assay measured the total plasma concentration of semaglutide (protein bound and unbound). The lower limit of quantification for semaglutide was 1.94 nmol/L. Blood samples for estimation of protein binding were collected 30 min before dosing. Semaglutide binding was determined using surface plasmon resonance biosensor technology (Biacore T100 Instrument, GE Healthcare Bio-Sciences AB, Umeå, Sweden), in which semaglutide was immobilized at the surface and diluted plasma, collected prior to dosing, was passed over the sensor chip. Binding was determined as the resultant change in the resonance angle of polarized light. The affinity (k d) and fraction unbound were determined using the 1:1 binding model.
2.5 Pharmacokinetic, Safety, and Statistical Analyses
Summary statistics and statistical analyses were based on all subjects exposed to a 0.5 mg dose of semaglutide. The six subjects who received a 10 µg/kg dose were evaluated for safety (reported AEs are described in Sect. 2.2), but were not included in the pharmacokinetics analysis set.
The primary pharmacokinetic endpoint was the area under the semaglutide plasma concentration–time curve (AUC) from time zero to infinity (AUC∞). Secondary pharmacokinetic endpoints included C max, time to reach C max following drug administration (t max), t ½, and apparent total clearance of the drug from plasma after subcutaneous administration (CL/F). In addition, for subjects in the ESRD and normal renal function groups, AUC from time zero to 48 h (AUC48) and AUC from 48 to 96 h (AUC48–96) were calculated.
The terminal elimination rate constant (λ z) was approximated by log-linear regression on the log-linear phase of the plasma concentration profile. AUC was calculated using the trapezoidal rule, and AUC∞ was then extrapolated from the time of last quantifiable observation (t) to infinite time using AUC( t–∞) = C(t)/λ z. The t ½ was defined as ln2/λ z and CL/F was estimated as dose/AUC∞.
To assess the effect of RI on semaglutide pharmacokinetic parameters, AUC∞ was log-transformed and analyzed using an analysis of covariance (ANCOVA) model, with renal group as a fixed effect. In addition, because of differences in baseline characteristics between the groups, the analysis was performed post hoc with age, sex, and log(weight) as explanatory variables. A two-stage procedure was pre-specified: in Stage 1 exposure was considered equivalent between the severe RI group and the normal renal function group if the 95% confidence intervals (CIs) for the ratio of AUC∞ (unadjusted) were contained within the pre-specified ‘no effect’ interval of 0.70–1.43 (both inclusive). The ‘no effect’ boundaries were selected based on the observation that semaglutide, as a GLP-1 analog, displays a wide therapeutic index. If equivalence was not shown in Stage 1, only then was Stage 2 performed, where equivalence between the mild RI group and the normal renal function group, and between the moderate RI group and the normal renal function group, was tested. Data from the ESRD group were also analyzed regardless of Stage 1 outcome. Correction for multiplicity due to multiple testing was performed by using a CI at the 95% level for each stage instead of 90% and a hierarchical testing scheme for the comparisons in Stage 2 (mild vs. normal followed by moderate vs. normal). As Stage 2 was performed, further analysis of the association between CLCR and AUC∞ and CLCR and C max was carried out (as pre-specified) by linear regression of the log-transformed endpoint, with log-transformed CLCR as the independent variable. C max was analyzed in the same model as AUC∞.
Tolerability of semaglutide was assessed through AEs, physical examination, electrocardiogram, vital signs, hypoglycemic events, plasma glucose, and clinical laboratory parameters (biochemistry, hematology, and urinalysis). Treatment-emergent AEs (TEAEs) were defined as events occurring between the dosing day and the follow-up visit +14 days (i.e., up to 5 weeks post-dose, corresponding to five half-lives of semaglutide), or starting before the dosing day with increasing severity during the period.
Hypoglycemic episodes were defined as ‘major’, ‘minor’, or ‘symptoms only’: major if the subject was unable to self-treat, minor if the subject was able to self-treat and plasma glucose was <3.1 mmol/L (56 mg/dL), or symptoms only if the subject was able to self-treat and had either a plasma glucose measurement ≥3.1 mmol/L or no plasma glucose measurement available. Safety endpoints were summarized using descriptive statistics.
3 Results
3.1 Subject Demographics
Following the decision to reduce the semaglutide dose from 10 µg/kg to 0.5 mg, a total of 56 subjects were exposed to a single 0.5 mg dose of semaglutide; 55 subjects completed the study and one subject withdrew due to nausea. Of those entering the study, 14 had normal renal function, 11 had mild RI, 11 had moderate RI, ten had severe RI, and ten had ESRD requiring hemodialysis. The enrollment of subjects with normal renal function was aimed to be balanced with regard to overall age, sex, and body weight distribution of the subjects with RI (Table 1). However, subjects with normal renal function had, on average, a lower body weight and were older than subjects with ESRD, and had a higher body weight and were younger than subjects with mild to severe RI. All groups included male and female subjects but, overall, there were more males than females. Of the subjects with RI, nine had type 2 diabetes as a concomitant illness at baseline.
3.2 Pharmacokinetics
Mean semaglutide plasma concentration over time by renal group is shown in Fig. 2, and all pharmacokinetic endpoints for semaglutide are shown in Table 2. Geometric means [coefficient of variation (CV), %] for AUC∞ showed no consistent trend with decreasing renal function, ranging from a lowest exposure of 2567 nmol h/L (18%) in the ESRD group to a highest exposure of 3179 nmol h/L (22%) in the severe RI group. Subjects with severe RI had a 22% higher mean exposure of semaglutide than subjects with normal renal function and the ratio did not demonstrate equivalence according to the predefined criteria for 95% CI (0.70–1.43). The CIs for the ratios of AUC∞ for semaglutide in the mild and moderate RI and ESRD groups were within the ‘no effect’ interval and were considered equivalent to the normal renal function group. When adjusted for differences between groups in age, sex, and log(weight), all of the 95% CIs for the ratio of AUC∞ between each RI group and the normal renal function group were contained within the pre-specified interval (0.70–1.43; Table 2).

Mean semaglutide plasma concentration over time by renal function group after a single dose of semaglutide 0.5 mg. The study period was from Day 1, 0 h (dosing) to Day 21, 480 h (follow-up visit). ESRD end-stage renal disease
Mean C max for semaglutide was highest in subjects with normal renal function (10.3 nmol/L) and lowest in subjects with ESRD (7.4 nmol/L), whereas across the other groups mean C max was 9.0–9.8 nmol/L (Table 2). When adjusted for differences between groups in age, sex, and log(weight), C max was 10–20% lower in subjects with RI than in subjects with normal renal function, with no clear trend in severity of RI (Table 2). The t max varied across the groups (range 24–51 h); mean t ½ was longer in subjects with severe RI (221 h) and those with ESRD (243 h) than in subjects with mild and moderate RI or normal renal function.
Exposure to semaglutide (AUC∞) as a function of CLCR and C max as a function of CLCR in all subjects, following a single dose of 0.5 mg semaglutide, are shown in Fig. 3a, b, respectively. There was no linear relationship between CLCR across groups (normal, mild, moderate, severe, and ESRD) and semaglutide exposure (p = 0.127), or between CLCR across groups and semaglutide C max (p = 0.164). When adjusted for differences in age, sex, and body weight, the small observed slopes for AUC∞ and C max (approximately −0.06 and 0.08, respectively) were unchanged; however, the association between CLCR and AUC∞ was statistically significant (p = 0.041). A slope of −0.06 corresponds to an increase in exposure of 14% in subjects with a CLCR of 10 mL/min versus subjects with a CLCR of 90 mL/min.

a Exposure of semaglutide and b maximum semaglutide plasma concentration versus creatinine clearance following a single dose of semaglutide 0.5 mg by renal function group. AUC ∞ area under the plasma concentration–time curve from time zero to infinity, C max maximum plasma drug concentration, ESRD end-stage renal disease
The point estimate of the ratio (ESRD group/normal renal function group) for AUC48 (without hemodialysis) was 0.72 (90% CI 0.57–0.91) and for AUC48–96 (with hemodialysis) was 0.82 (90% CI 0.67–0.99). The estimated ratios were also comparable when adjusted for differences in age, sex, and body weight [0.81 (90% CI 0.66–1.01) for AUC48 and 0.93 (90% CI 0.78–1.10) for AUC48–96], indicating that hemodialysis did not appear to affect the pharmacokinetics of semaglutide.
There was no difference in protein binding between the groups; the fraction of unbound semaglutide across renal groups was low (<1%; data not shown). All subjects had normal albumin concentrations at baseline and throughout the study (except for one subject in the normal renal function group at the screening visit).
3.3 Adverse Events and Other Safety Assessments
The safety population comprised all 56 enrolled subjects who received semaglutide 0.5 mg. One subject in the mild RI group withdrew from the study on Day 2 because of nausea.
Overall TEAEs are shown in Table 3. In total, 38 subjects experienced 89 events with semaglutide 0.5 mg; none were serious or severe and the majority were mild. The AE profiles in the normal renal function and mild and moderate RI groups were similar; compared with these three groups, the proportion of subjects experiencing AEs was higher in the severe RI group and lower in the ESRD group. The most frequently reported TEAEs were gastrointestinal (a total of 35 subjects experienced 61 events), with nausea being the most frequent (41% of subjects). A higher proportion of subjects experienced nausea in the severe group than in all other groups. For AEs in the six subjects who received a single dose of semaglutide 10 µg/kg, see Sect. 2.2.
One subject with severe RI (who did not have type 2 diabetes, but had experienced nausea, vomiting, and decreased appetite from the dosing day) had two major hypoglycemic events, one subject with moderate RI and one with ESRD experienced minor hypoglycemia, and two subjects in the moderate RI group experienced symptoms-only hypoglycemia.
No clinically significant values or appreciable changes in vital signs (blood pressure and pulse), electrocardiogram, physical examinations, or clinical laboratory assessments were observed.
4 Discussion
We examined the human GLP-1 analog semaglutide in subjects with normal renal function and those with varying degrees of RI, to investigate whether the pharmacokinetics change across groups and to determine whether dose adjustments are needed. The study population included a broad range of subjects with varying severities of RI: mild, moderate, severe, and a group with ESRD undergoing regular hemodialysis.
The results of this study demonstrate that, for moderate or mild RI, or in subjects with ESRD, the exposure of semaglutide is within the ‘no effects’ limits, compared with the normal renal function group. The comparison between the group with normal renal function and the group with severe RI did not demonstrate equivalence (the 95% CI exceeded the ‘no effect’ limits), and the mean semaglutide exposure was 22% higher in the severe RI group. C max for the ESRD group was outside the pre-specified ‘no effects’ limits and appeared to be lower than in the other RI groups. However, baseline characteristics of subjects with normal renal function were somewhat different from those in the four RI groups. On average, subjects with normal renal function had a higher body weight and were younger (84.9 kg, 54.6 years) than subjects in the mild, moderate, and severe RI groups (78.1–80.1 kg, 62.8–66.5 years) but had a lower body weight and were older than subjects with ESRD (97.2 kg, 48.2 years). When the data were adjusted for differences in age, sex, and body weight among the groups, all comparisons for exposure were within the ‘no effect’ limits, and C max was similar between RI and ESRD groups, which were all 10–20% lower than the normal renal function group (Table 2). In addition, there was no relationship between CLCR across RI groups (normal, mild, moderate, severe, and ESRD) and semaglutide exposure, or between the CLCR and semaglutide C max. The pharmacokinetics of semaglutide did not appear to be affected by hemodialysis, as the semaglutide exposure ratio of ESRD subjects to healthy subjects during dialysis (AUC48–96) was similar to the ratio obtained when subjects were not receiving dialysis (AUC48).
Semaglutide (single dose of 0.5 mg) was well-tolerated, despite not being administered with a dose-escalation regimen. As expected, the AEs were mainly gastrointestinal and, while one subject withdrew from the study because of nausea, the majority of events were mild, and all were mild to moderate, in severity. While this was a single-dose study, the semaglutide dose-escalation regimens used in two other studies—Kapitza et al. [21] (starting dose 0.25 mg for 4 weeks, then 0.5 mg for 4 weeks and 1.0 mg for 5 weeks) and Nauck et al. [18] (0.4 mg for 1 week followed by 0.8 mg for 11 weeks)—reduced the incidence of nausea, indicating tolerance development compared with no or weekly dose escalation. The starting dose and dose-escalation regimen reported by Kapitza et al. [21] has been used in the semaglutide phase III development program, in which semaglutide showed a gastrointestinal AE profile comparable to that of other GLP-1 receptor agonists [23]. As the C max in the RI groups was not higher than in the normal renal function group—indeed, when adjusted for age, sex and body weight, C max was 10–20% lower than in the normal group—no tolerability issues are expected in clinical practice.
In this study, two major episodes of hypoglycemia were reported in a subject with severe RI (who did not have type 2 diabetes). This may be treatment related or could be linked to the presence of RI; in a retrospective cohort analysis of 243,222 patients, Moen et al. [14] showed that chronic renal disease is a significant risk factor for hypoglycemia in patients with or without diabetes. No other safety or tolerability concerns were raised with a single dose of semaglutide 0.5 mg, and none of the TEAEs were classified as serious or severe.
This study was conducted according to regulatory standards comprising four RI groups (mild, moderate and severe RI, and ESRD), with ≥10 subjects in each group and a comparable reference group with normal renal function [33, 34]; the results indicate that the pharmacokinetic properties of semaglutide are not significantly affected by renal function. Therefore, specific dose adjustment may not be warranted in subjects with RI. Our study is supported by a recent trial reporting the absorption, metabolism, and excretion of a single subcutaneous 0.5 mg dose of [3H]-radiolabeled semaglutide in healthy male subjects, showing that semaglutide has very limited renal excretion in its intact form [22].
Other available GLP-1 receptor agonists have been approved for use in mild and moderate RI [35,36,37,38,39,40]. These drugs are not recommended for patients with severe RI, ESRD, or who are on dialysis because of limited therapeutic experience [30, 35]. Subjects with a broad variety of RI were enrolled in the Semaglutide Unabated Sustainability in Treatment of Type 2 Diabetes (SUSTAIN) program (the phase III development program for semaglutide).
Recently, findings from the SUSTAIN 6 (Trial to Evaluate Cardiovascular and Other Long-term Outcomes with Semaglutide in Subjects with Type 2 Diabetes) cardiovascular outcomes study—in which more than two-thirds of patients had some degree of RI (estimated glomerular filtration rate <90 mL/min/1.73 m2)—showed that treatment with semaglutide (on a background of standard of care) resulted in significant and sustained reductions in glycated hemoglobin (HbA1c) versus placebo (mean HbA1c was 0.7 and 1.0 percentage points lower with semaglutide 0.5 and 1.0 mg, respectively, vs. placebo, both p < 0.001). In the SUSTAIN 6 study, gastrointestinal AEs were more frequent with semaglutide than with placebo, but most were mild or moderate in severity [23]. In a subgroup analysis for the primary outcome (first occurrence of cardiovascular death, non-fatal myocardial infarction, or non-fatal stroke), no significant treatment interactions were identified for the renal function subgroups analyzed [23]. Therefore, SUSTAIN 6 showed that the effects of semaglutide on the primary composite endpoint were not affected by renal subgroup [23].
5 Conclusion
When adjusted for differences in sex, age, and body weight, semaglutide exposure is not affected by RI. Therefore, semaglutide appears to be a useful treatment option for patients with type 2 diabetes who also have impaired renal function, including those with ESRD undergoing hemodialysis. Dose adjustment may not be warranted for patients with RI.
References
Cavanaugh K. Diabetes management issues for patients with chronic kidney disease. Clin Diabet. 2007;25(3):90–7.
de Boer IH, Rue TC, Hall YN, Heagerty PJ, Weiss NS, Himmelfarb J. Temporal trends in the prevalence of diabetic kidney disease in the United States. JAMA. 2011;305(24):2532–9.
Pyram R, Kansara A, Banerji MA, Loney-Hutchinson L. Chronic kidney disease and diabetes. Maturitas. 2012;71(2):94–103.
Assogba GF, Couchoud C, Roudier C, Pornet C, Fosse S, Romon I, et al. Prevalence, screening and treatment of chronic kidney disease in people with type 2 diabetes in France: the ENTRED surveys (2001 and 2007). Diabetes Metab. 2012;38(6):558–66.
Middleton RJ, Foley RN, Hegarty J, Cheung CM, McElduff P, Gibson JM, et al. The unrecognized prevalence of chronic kidney disease in diabetes. Nephrol Dial Transplant. 2006;21(1):88–92.
New JP, Middleton RJ, Klebe B, Farmer CK, de Lusignan S, Stevens PE, et al. Assessing the prevalence, monitoring and management of chronic kidney disease in patients with diabetes compared with those without diabetes in general practice. Diabet Med. 2007;24(4):364–9.
Metsarinne K, Broijersen A, Kantola I, Niskanen L, Rissanen A, Appelroth T, et al. High prevalence of chronic kidney disease in Finnish patients with type 2 diabetes treated in primary care. Prim Care Diabetes. 2015;9(1):31–8.
Van Buren PN, Toto R. Hypertension in diabetic nephropathy: epidemiology, mechanisms, and management. Adv Chronic Kidney Dis. 2011;18(1):28–41.
Iglesias P, Diez JJ. Insulin therapy in renal disease. Diabetes Obes Metab. 2008;10(10):811–23.
McGill JB. Anti-diabetes therapy: safety considerations for patients with impaired kidney function. Postgrad Med. 2014;126(3):161–71.
Dejager S, Schweizer A. Incretin therapies in the management of patients with type 2 diabetes mellitus and renal impairment. Hosp Pract (1999). 2012;40(2):7–21.
Giorda CB, Nada E, Tartaglino B. Pharmacokinetics, safety, and efficacy of DPP-4 inhibitors and GLP-1 receptor agonists in patients with type 2 diabetes mellitus and renal or hepatic impairment. A systematic review of the literature. Endocrine. 2014;46(3):406–19.
Davis TM. Dipeptidyl peptidase-4 inhibitors: pharmacokinetics, efficacy, tolerability and safety in renal impairment. Diabetes Obes Metab. 2014;16(10):891–9.
Moen MF, Zhan M, Hsu VD, Walker LD, Einhorn LM, Seliger SL, et al. Frequency of hypoglycemia and its significance in chronic kidney disease. Clin J Am Soc Nephrol. 2009;4(6):1121–7.
Muhlhauser I, Toth G, Sawicki PT, Berger M. Severe hypoglycemia in type I diabetic patients with impaired kidney function. Diabetes Care. 1991;14(4):344–6.
Farrokhi F, Klindukhova O, Chandra P, Peng L, Smiley D, Newton C, et al. Risk factors for inpatient hypoglycemia during subcutaneous insulin therapy in non-critically ill patients with type 2 diabetes. J Diabetes Sci Technol. 2012;6(5):1022–9.
Arnouts P, Bolignano D, Nistor I, Bilo H, Gnudi L, Heaf J, et al. Glucose-lowering drugs in patients with chronic kidney disease: a narrative review on pharmacokinetic properties. Nephrol Dial Transplant. 2014;29(7):1284–300.
Nauck MA, Petrie JR, Sesti G, Mannucci E, Courreges JP, Lindegaard ML, et al. A phase 2, randomized, dose-finding study of the novel once-weekly human GLP-1 analog, semaglutide, compared with placebo and open-label liraglutide in patients with type 2 diabetes. Diabetes Care. 2016;39(2):231–41.
Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132(6):2131–57.
Deacon CF. Circulation and degradation of GIP and GLP-1. Horm Metab Res. 2004;36(11–12):761–5.
Kapitza C, Nosek L, Jensen L, Hartvig H, Jensen CB, Flint A. Semaglutide, a once-weekly human GLP-1 analog, does not reduce the bioavailability of the combined oral contraceptive, ethinylestradiol/levonorgestrel. J Clin Pharmacol. 2015;55(5):497–504.
Jensen L, Helleberg H, Roffel A, van Lier J, Bjørnsdottir I, Pedersen PJ, et al. Absorption, metabolism and excretion of the GLP-1 analogue semaglutide in humans and nonclinical species. Eur J Pharm Sci. 2017. doi:10.1016/j.ejps.2017.03.020.
Marso SP, Bain SC, Consoli A, Eliaschewitz FG, Jodar E, Leiter LA, et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016;375(19):1834–44.
Young MA, Wald JA, Matthews JE, Yang F, Reinhardt RR. Effect of renal impairment on the pharmacokinetics, efficacy, and safety of albiglutide. Postgrad Med. 2014;126(3):35–46.
Jacobsen LV, Hindsberger C, Robson R, Zdravkovic M. Effect of renal impairment on the pharmacokinetics of the GLP-1 analogue liraglutide. Br J Clin Pharmacol. 2009;68(6):898–905.
Hermansen K, Kolotkin RL, Hammer M, Zdravkovic M, Matthews D. Patient-reported outcomes in patients with type 2 diabetes treated with liraglutide or glimepiride, both as add-on to metformin. Prim Care Diabetes. 2010;4(2):113–7.
Mikhail NE. Is liraglutide a useful addition to diabetes therapy? Endocr Pract. 2010;16(6):1028–37.
Linnebjerg H, Kothare PA, Park S, Mace K, Reddy S, Mitchell M, et al. Effect of renal impairment on the pharmacokinetics of exenatide. Br J Clin Pharmacol. 2007;64(3):317–27.
Davies MJ, Bain SC, Atkin SL, Rossing P, Scott D, Shamkhalova MS, et al. Efficacy and safety of liraglutide versus placebo as add-on to glucose-lowering therapy in patients with type 2 diabetes and moderate renal impairment (LIRA-RENAL): a randomized clinical trial. Diabetes Care. 2016;39(2):222–30.
Davies M, Chatterjee S, Khunti K. The treatment of type 2 diabetes in the presence of renal impairment: what we should know about newer therapies. Clin Pharmacol. 2016;8:61–81.
Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31–41.
European Medicines Agency. ICH topic E6 (R1). Guideline for good clinical practice. 2002. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002874.pdf. Accessed 2 Dec 2016.
European Medicines Agency. Note for guidance on the evaluation of the pharmacokinetics of medicinal products in patients with impaired renal function. 2004. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003123.pdf. Accessed 2 Dec 2016.
US Food and Drug Administration. Guidance for industry. Pharmacokinetics in patients with impaired renal function – study design, data analysis and impact on dosing and labeling. 1998. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm072127.pdf. Accessed 2 Dec 2016.
European Medicines Agency. Victoza summary of product characteristics. 2016. https://www.medicines.org.uk/emc/medicine/21986. Accessed 2 Dec 2016.
European Medicines Agency. Eperzan summary of product characteristics. 2016. https://www.medicines.org.uk/emc/medicine/31399. Accessed 2 Dec 2016.
European Medicines Agency. Trulicity summary of product characteristics. 2016. https://www.medicines.org.uk/emc/medicine/29747. Accessed 2 Dec 2016.
European Medicines Agency. Bydureon summary of product characteristics. 2016. https://www.medicines.org.uk/emc/medicine/24665. Accessed 2 Dec 2016.
European Medicines Agency. Byetta summary of product characteristics. 2016. https://www.medicines.org.uk/emc/medicine/19257. Accessed 2 Dec 2016.
European Medicines Agency. Lyxumia summary of product characteristics. 2016. https://www.medicines.org.uk/emc/medicine/27405. Accessed 2 Dec 2016.
Acknowledgments
The authors would like to thank all subjects who participated in this study. In addition, the authors thank Novo Nordisk A/S for their contribution to the study design, data management, and data analysis, Sayeh Tadayon (Novo Nordisk) for her review and input to the manuscript, and Flavia Sciota, PhD (AXON Communications) for medical writing and editorial assistance. All contributors received compensation from Novo Nordisk.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by Novo Nordisk A/S.
Conflicts of interest
The study was sponsored by Novo Nordisk A/S, the manufacturer of semaglutide. AF, JBJ, and JDK are employees of, and own shares in, Novo Nordisk A/S. TCM is an employee of Orlando Clinical Research Center.
Ethical Approval
The study was conducted in accordance with Good Clinical Practice, the Declaration of Helsinki, and current guidelines for studies in patients with RI.
Informed Consent
All participants gave their written consent prior to the start of any study-related activities.
Author Contributions
TCM conducted the study and collected the data. TCM, AF, JBJ, JDK, and KL performed the data analysis, wrote, and revised the manuscript. All authors gave final approval of the manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Marbury, T.C., Flint, A., Jacobsen, J.B. et al. Pharmacokinetics and Tolerability of a Single Dose of Semaglutide, a Human Glucagon-Like Peptide-1 Analog, in Subjects With and Without Renal Impairment. Clin Pharmacokinet 56, 1381–1390 (2017). https://doi.org/10.1007/s40262-017-0528-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-017-0528-2