
Abstract
Background
Insulin degludec is a new-generation basal insulin with an ultra-long duration of action. We evaluated the pharmacokinetic properties of insulin degludec in subjects with normal renal function; mild, moderate or severe renal impairment; or end-stage renal disease (ESRD) undergoing hemodialysis.
Methods
Thirty subjects (n = 6 per group) received a single subcutaneous dose of 0.4 U/kg insulin degludec. Blood samples up to 120 h post-dose and fractionated urine samples were collected.
Results
The ultra-long pharmacokinetic properties of insulin degludec were preserved in subjects with renal impairment, with no statistically significant differences in absorption or clearance, compared with subjects with normal renal function. In subjects with ESRD, pharmacokinetic parameters were similar whether the insulin degludec pharmacokinetic assessment period included hemodialysis or not, and total exposure was comparable to subjects with normal renal function. Simulated mean steady-state pharmacokinetic profiles were comparable between groups.
Conclusion
This study indicated dose adjustments due to impaired renal function should not be required for insulin degludec.
Similar content being viewed by others

1 Introduction
Impaired renal function is an increasingly common long-term complication of diabetes mellitus [1, 2]. Prevalence rates of co-morbid chronic kidney disease are approximately 30 % in subjects with diabetes [3–5], and type 2 diabetes has now been identified as the most common cause of end-stage renal disease (ESRD) [6].
As exogenous insulin is primarily cleared via renal metabolism [1, 7–9], impaired renal function in patients with diabetes can result in decreased clearance of insulin and, consequently, prolonged exposure [10]. Diabetes patients with impaired renal function may therefore be at increased risk of hypoglycemia [9, 11–13]. In addition, deterioration of renal function can lead to increased exposure to insulin therapy, potentially increasing the risk of hypoglycemia and hampering effective diabetes management in these patients [1].
Impairments in renal function in patients with diabetes have been shown to affect the pharmacokinetics of some insulin products [14], including regular human insulin and insulin lispro [15, 16]. However, the pharmacokinetics of other insulin products, including insulin aspart [17, 18] and the long-acting insulin product insulin detemir [19, 20], are reportedly unaffected, and it has been suggested that insulin analogs may maintain their pharmacokinetic profiles in patients with renal failure [7].
Insulin degludec (Tresiba®, Novo Nordisk A/S) is a new-generation basal insulin with a distinct absorption mechanism that results in an ultra-long duration of action. Upon subcutaneous injection, insulin degludec forms long chains of multi-hexamers, resulting in a soluble depot in the subcutaneous tissue from which insulin degludec monomers gradually separate [21, 22]. This results in slow and continuous absorption of insulin degludec into the circulation. Insulin degludec has a flat and stable glucose-lowering effect [23], with a four-times lower within-subject variability in glucose-lowering effect [area under the curve (AUC) values] than insulin glargine [24]. These properties of insulin degludec lead to a decreased risk of hypoglycemia compared with insulin glargine [24–28].
The aim of the present study was to determine whether the ultra-long pharmacokinetic properties of insulin degludec observed in subjects with normal renal function are preserved in those with renal impairment. We evaluated pharmacokinetic parameters following a single dose of insulin degludec in subjects with mild, moderate, or severe renal impairment and subjects with ESRD undergoing dialysis treatment, and compared these with parameters measured in subjects with normal renal function. The tolerability profile of insulin degludec after a single dose was also assessed in these subjects. In addition, mean steady-state pharmacokinetic profiles were simulated based on single-dose pharmacokinetic profiles.
2 Methods
2.1 Study Populations
Study participants were men and women aged 18–85 years with a body mass index of ≤40 kg/m2. Subjects had normal renal function or stable renal impairment, according to predefined creatinine clearance (CLCR) criteria estimated according to the Cockcroft and Gault formula, during the screening visit. Accordingly, subjects were divided into the following groups: normal renal function (CLCR >80 mL/min), mild (CLCR 50–80 mL/min), moderate (CLCR 30–49 mL/min), or severe (CLCR <30 mL/min) renal impairment, or subjects with ESRD requiring hemodialysis. Inclusion criteria permitted impaired glucose tolerance or diabetes in subjects with renal impairment.
Exclusion criteria for all subjects included known or suspected allergy to the trial product or related products; the presence of any clinically significant abnormality in hematology, biochemistry or urinalysis (when accounting for the underlying disease); current hepatic dysfunction or severe hepatic disease during the previous 12 months; impaired hepatic function, defined as hepatic enzyme elevation ≥2.5 times upper limit of normal; peritoneal dialysis and renal transplantation. For subjects with normal renal function, exclusion criteria also included the use of agents known to alter tubular secretion of creatinine (within 14 days or five half-lives prior to first dosing), monoamine oxidase inhibitors, thyroid hormones (unless the use of these was stable 3 months prior to trial initiation), or growth hormone.
2.2 Study Design and Pharmacokinetic Sampling
This was a single-center (Department of Clinical Pharmacology, St Imre Teaching Hospital, Budapest, Hungary), single-dose, open-label, parallel-group trial (ClinicalTrials.gov number NCT01006057). A sample size of six subjects in each group was used to meet the primary objective of this trial and to adhere to current guidelines from the US FDA and the European Medicines Agency [29, 30].
Before trial initiation, the protocol and amendments, subject consent form, and information sheet were approved by the National Institute for Pharmacy of the National Institute for Quality- and Organizational Development in Healthcare and Medicine (GYEMSZI-OGYI), Hungary, and by the independent Scientific and Research Ethics Committee of the Medical Research Council, Hungary (ETT-TUKEB). The study was performed in accordance with the Declaration of Helsinki, and its amendments, and Good Clinical Practice as defined by the International Conference on Harmonisation. Subjects were informed of the risks and benefits of the trial and that they could withdraw from the trial at any time. Consent was obtained in writing before any trial-related activities commenced; the investigator retained the consent forms.
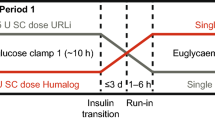
Following screening (visit 1) subjects were allocated to one of five groups, according to their renal function. Subjects allocated to non-ESRD groups received a single dose of insulin degludec during visit 2 (2–21 days after visit 1), administered at 0800 hours. Subjects allocated to the ESRD group received two single doses of insulin degludec, each administered at separate dosing visits (visit 2, 2–21 days after visit 1; and visit 3, 13–21 days after visit 2). The first dose of insulin degludec was administered at 1200 hours, immediately after completion of a hemodialysis session (visit 2), while the second dose of insulin degludec was administered at 1900 hours (visit 3), approximately 13 h before the start of a hemodialysis session, in order that hemodialysis was conducted at approximately the time that predicted insulin degludec t max [time at which maximum serum insulin degludec concentration (C max)] is achieved (data on file). Insulin degludec was provided in 3 mL Penfill® cartridges (100 U/mL) for dosing and was administered subcutaneously as a single dose of 0.4 U/kg into a lifted skin fold on the anterior surface of the thigh. Subjects remained in-house for a minimum of 48 h following dosing, or longer if deemed necessary by the investigator. A follow-up visit (visit 3 in non-ESRD subjects; visit 4 in ESRD subjects) was conducted 7–21 days after the final dosing in all subjects (Fig. 1). The total study duration was 10–43 days for non-ESRD subjects and 23–64 days for ESRD subjects.

Trial design for non-ESRD subjects with various degrees of renal impairment, and ESRD subjects. ESRD end-stage renal disease
In all subjects, blood samples were collected pre-dose for blood glucose analysis and insulin degludec pharmacokinetic analysis, post-dose at intervals of 0.5, 1, 2, 4, 6 h and then hourly until 20 h post-dose, then at 22, 24, 30, 36 and 48 h post-dose for pharmacokinetic analysis. Blood glucose analysis was performed approximately every 30 min from 0.5 to 19 h, and then at 20, 22, 24, 30, 36, and 48 h post-dose. Additional samples were taken at 72, 96, and 120 h for pharmacokinetic analysis and blood glucose analysis after dosing in non-ESRD subjects, and at 68 h post-dose in ESRD subjects. In all subjects, a baseline urine sample was collected at 0 h (pre-dose), and fractionated urine collection was performed to determine insulin degludec concentration/excretion after insulin degludec administration at predefined intervals post-dose at visit 2 (0–8, 8–16, and 16–24 h). A dialysate sample was collected for pharmacokinetic analysis from ESRD subjects during the 4-h dialysis session at the second dosing visit (visit 3) at 0.5, 1.5, 2.5, and 3.5 h.
2.3 Assay Methodology
Serum, urine, and dialysate concentrations of insulin degludec were measured using a validated, sandwich enzyme-linked immunosorbent assay (ELISA), specific for insulin degludec with a lower limit of quantification (LLOQ) of 20 pM for serum and urine and 100 pM for dialysate. For the assay, the capture antibody was a mouse monoclonal antibody specific for human insulin (HUI 001) and the detection antibody was a biotin-labelled monoclonal mouse antibody (NN454-1 F31) [23].
2.4 Data and Statistical Analysis
The primary pharmacokinetic endpoint, calculated in non-ESRD subjects, was the area under the insulin degludec serum concentration–time curve from 0 to 120 h, following a single dose (AUC0–120h). AUC0–120h was derived using the linear trapezoidal technique based on observed values and actual measurement times between 0 and 120 h, with missing values interpolated.
Secondary pharmacokinetic endpoints, calculated in all subjects, included the area under the insulin degludec serum concentration–time curve from zero to infinity, following a single dose (AUC0–∞), maximum serum insulin degludec concentration and insulin degludec apparent clearance (CL/F). Secondary pharmacokinetic endpoints calculated only in ESRD subjects included the area under the serum insulin degludec concentration–time curve from 13 to 17 h, following a single dose (AUC13–17h). Secondary pharmacokinetic endpoints were derived from individual serum insulin degludec concentration–time curves. CL/F was calculated as dose/AUC0–∞. In ESRD subjects, pharmacokinetic endpoints were calculated following a single dose of insulin degludec administered either at the end of a hemodialysis session or 13 h before a hemodialysis session.
To assess the effect of the degree of renal impairment on insulin degludec pharmacokinetic parameters for non-ESRD subjects, AUC0–120h, C max and CL/F were log-transformed and analyzed using an analysis of variance (ANOVA) model with log CLCR at screening, and sex and age at baseline as fixed effects. To assess the effect of hemodialysis on CL/F of insulin degludec for ESRD subjects, CL/F was log-transformed and analyzed using an ANOVA model, with visit as fixed effect and subject as random effect. Pharmacokinetic endpoints were summarized using descriptive statistics.
Tolerability of insulin degludec was assessed through adverse events, physical examination, vital signs, electrocardiogram, hypoglycemic events and clinical laboratory tests (biochemistry, hematology and urinalysis). Adverse events were classified as mild, moderate, or severe, and as having a probable, possible, or unlikely relationship to the trial product by the investigator. Hypoglycemic episodes were defined as ‘confirmed’ if verified by a plasma glucose concentration <3.1 mmol/L (56 mg/dL), irrespective of symptoms, or classified as ‘severe’ (requiring assistance) as defined by American Diabetes Association guidelines [31]. Tolerability endpoints were summarized using descriptive statistics.
To simulate the mean steady-state pharmacokinetic profile of insulin degludec from this single-dose study, a population pharmacokinetic model was used. The model consisted of an absorption component and a disposition component. The absorption component had a depot compartment, a delay compartment, an absorption rate parameter, and a delay rate parameter. The disposition component had one compartment, a clearance parameter and a volume of distribution parameter. The parameters of the model were estimated in a population pharmacokinetic setting using a non-linear mixed-effects approach, which allowed individual sets of the four parameters for each of the subjects included in the trial to be obtained. The model was estimated in NONMEM version 7.1.2 installed in a validated computer environment. The values of the absorption rate parameter were subsequently calibrated based on information from the comprehensive clinical pharmacology programme conducted with insulin degludec. The same calibration factor was applied for all subjects. Using the individual parameters, a simulation of multiple dosing was carried out to obtain a mean steady-state profile. More specifically, multiple dosing for 6 days at a dose level of 0.4 U/kg was simulated by extrapolating the profile for each of the subjects and subsequently calculating the mean of the profiles on day 6.
3 Results
3.1 Subjects
A total of 48 subjects were screened, of which 16 were excluded (14 did not meet eligibility criteria and two withdrew consent). Thirty-two subjects met eligibility criteria and received at least one injection of insulin degludec. These subjects comprised the population for assessment of tolerability. Of these, two subjects within the normal renal function group were later discovered not to meet eligibility criteria for that group and were excluded. Thirty subjects therefore comprised the full analysis set; six subjects were included in each of the five renal function groups [normal renal function, mild renal impairment, moderate renal impairment or severe renal impairment (these four groups were termed non-ESRD subjects) and ESRD] (Fig. 1).
Subject demographics at baseline were largely comparable across renal function groups (Table 1). Subjects were White men and women with mean ages ranging from 57 to 72 years (Table 1). Eleven subjects with renal impairment also had type 2 diabetes as a concomitant illness at baseline and eight of these subjects were treated with insulin (Table 1). The mean duration of diabetes was 21 years.
3.2 Pharmacokinetics
Exposure to insulin degludec (AUC0–120h) in non-ESRD subjects following a single 0.4 U/kg dose is shown in Fig. 2 as a function of CLCR. AUC0–120h was not significantly affected by the degree of renal impairment (p = 0.26) following a single dose (Table 2). Geometric means [coefficient of variation (CV), %] for AUC0–∞ for the normal renal function group and the mild, moderate, and severe renal impairment groups are given in Table 3. The degree of renal impairment did not appear to have a significant effect on maximum serum insulin degludec concentration (C max) (p = 0.16) or apparent insulin degludec clearance (CL/F) [p = 0.29] following a single dose (Tables 2 and 3). The estimated ratios of AUC0–∞ and C max for insulin degludec in subjects with renal impairment versus subjects with normal renal function are given in Table 4. In 23 of the 30 subjects, concentrations of insulin degludec in the urine were consistently below the assay LLOQ for all four time intervals (pre-dose, and 0–8, 8–16, and 16–24 h post-dose). In four subjects (one with normal renal function, one with mild renal impairment, one with moderate renal impairment, and one with ESRD), urine insulin degludec concentration was marginally above the assay LLOQ for one time interval; in two subjects (one with mild renal impairment, one with moderate renal impairment), urine insulin degludec concentration was marginally above the assay LLOQ for two time intervals, and in one subject (with ESRD) urine insulin degludec concentration was above the assay LLOQ for three time intervals. Altogether, these data suggest minimal renal clearance of insulin degludec.

Total exposure of insulin degludec vs. creatinine clearance following a single dose of insulin degludec (0.4 U/kg subcutaneously). AUC 0–120h area under the insulin degludec serum concentration–time curve from 0 to 120 h
In the subjects with ESRD, the geometric means for AUC0–∞ were largely comparable irrespective of whether dosing occurred immediately after (see Table 3) or 13 h before [geometric mean (CV, %) 90,846 (26) pmol·h/L] hemodialysis. The maximum insulin degludec serum concentrations (C max) were also similar in ESRD subjects irrespective of whether dosing occurred immediately after (see Table 3) or 13 h before [2,955 (45) pmol/L] hemodialysis. Both AUC0–∞ and C max in ESRD subjects were comparable with the results obtained in the other groups (see Table 3).
To investigate further the effect of hemodialysis on the pharmacokinetic profile of insulin degludec, the AUC13–17h was calculated. This time interval corresponds to the 4-h period where hemodialysis occurred at the second dosing visit. AUC13–17h was 10,422 (32) and 10,114 (46) pmol·h/L for doses administered immediately after or 13 h before a hemodialysis session, respectively. This was to confirm that hemodialysis does not affect the pharmacokinetic profile of insulin degludec. There was no statistically significant effect of hemodialysis on CL/F (mean ratio before/after dialysis 1.23, 95 % CI 0.92–1.66; p = 0.13). Mean afferent and efferent concentrations of insulin degludec (i.e. concentrations in blood entering and leaving the dialysis apparatus during hemodialysis) were similar (data not presented), and all concentrations of insulin degludec in the dialysate were below the assay LLOQ, suggesting minimal clearance of insulin degludec during hemodialysis.
3.2.1 Simulated Mean Steady-State Insulin Degludec Concentrations
To simulate mean steady-state insulin degludec pharmacokinetic profiles for once-daily 0.4 U/kg subcutaneous administration, a population pharmacokinetic model was used. Profiles showed an even distribution of exposure across a 24-h dosing interval, regardless of renal function status (Fig. 3).

Simulated mean insulin degludec (IDeg) concentrations at steady state (IDeg 0.4 U/kg subcutaneously)
3.3 Tolerability
Three mild (injection site reaction, nausea and vomiting) and one moderate (vomiting) treatment-emergent adverse events were reported in three subjects treated with insulin degludec. Of these, only the injection site reaction, which was deemed mild in severity, was considered probably related to insulin degludec. None of the adverse events reported were considered severe.
One treatment-emergent confirmed hypoglycemic episode was reported in the study, in an ESRD subject with diabetes who had received insulin degludec 13 h before hemodialysis. No severe hypoglycemic episodes were reported. No clinically significant changes were observed in laboratory parameters, vital signs, physical examination findings, or electrocardiogram results for the duration of the study.
4 Discussion
This study demonstrated that the pharmacokinetic properties of insulin degludec were preserved in subjects with mild, moderate, or severe renal impairment and in subjects with ESRD, compared with subjects with normal renal function. Notably, the single-dose total exposure and maximum concentration as well as clearance of insulin degludec were comparable for subjects with normal and varying degrees of impaired renal function. Furthermore, renal clearance of insulin degludec was shown to be negligible and hemodialysis did not significantly affect the clearance. The initial peptide cleavage of insulin degludec occurs within the cell and is the same as observed for human insulin [32]. The fatty-acid side chain is extensively metabolized similarly to other naturally occurring fatty acids [32]. Simulated mean steady-state pharmacokinetic profiles for insulin degludec in the present study supported the conclusion that the long, flat, consistent exposure of insulin degludec across one dosing interval is preserved among groups regardless of renal function.
Insulin degludec was well tolerated in this study. Few adverse events (four events in three subjects) were reported during the trial and there were no obvious differences in the frequency of adverse events between groups. One confirmed hypoglycemic episode occurred and no severe episodes were reported. The episode of hypoglycemia occurred in a subject with diabetes with ESRD who was not receiving any other antidiabetic therapy. Due to the single-dose design of this trial using a fixed-dose level, any observations on hypoglycemic episodes may not be clinically relevant. A pre-planned meta-analysis of the insulin degludec phase IIIa trial program has demonstrated lower rates of overall confirmed and nocturnal hypoglycemia with the use of insulin degludec, compared with insulin glargine, in subjects with type 2 diabetes [27].
Renal impairment is known to affect insulin clearance [7]; however, the effects of renal impairment on the pharmacokinetics of individual insulin products vary. To date, only a few studies have been conducted to assess the effect of renal impairment on the pharmacokinetics of basal insulin analogs [14]. A study assessing the effects of a regimen of insulin glargine and insulin glulisine reported the use of dose reduction to decrease the risk of hypoglycemia in subjects with renal impairment [33], while another study demonstrated that hemodialysis did not alter insulin glargine dosage requirements in ESRD patients [34]. The summary of product characteristics (SmPC) for insulin glargine does not offer specific recommendations for dosing in subjects with renal impairment, but states that insulin requirements may be diminished in such patients [35]. A similar statement, that insulin requirements may be altered or diminished in patients with renal impairment, is made in the SmPCs of human insulin products [36–38]. The pharmacokinetics of long-acting insulin detemir are not affected by renal impairment [20]. This is reflected in the SmPC for insulin detemir, which states that impairments in renal function do not lead to any clinically relevant differences in the pharmacokinetics of this product [19]. Similarly, the overall findings of this study suggest that specific dose adjustment is not required for the administration of insulin degludec in subjects with impaired renal functioning, compared to those with normal renal function. In addition to these findings, a study examining the effects of varying degrees of impaired hepatic function on the ultra-long pharmacokinetics of insulin degludec found that these properties were also preserved in subjects with hepatic impairment [39].
As with any other study, this trial has its strengths and limitations. Importantly, the trial was conducted according to regulatory standards having four renal impairment groups (mild, moderate, severe renal impairment and ESRD) with six subjects in each group, and with a control group comparable to the impaired renal function groups with respect to age, sex and weight [29, 30]. Nevertheless, the small size of this study should be borne in mind, and insulin should always be titrated according to individual patient requirements.
Ideally, all subjects would have been recruited from the intended patient population; however, as there is no indication of any difference in pharmacokinetic properties of insulin degludec between subjects with diabetes and healthy subjects, and to make recruitment feasible, both subjects with and without diabetes were allowed to participate. In contrast to the current single-dose study, a multiple-dose study with insulin degludec using a clinically relevant maintenance dose would not have been feasible as non-diabetic subjects could not be included for safety reasons (due to the risk of hypoglycemia). Therefore, to address the steady-state pharmacokinetics for insulin degludec in subjects with renal impairment, we simulated the pharmacokinetic profile of insulin degludec at steady state using a population pharmacokinetic model based on data from the current trial. The outcome suggests that at steady state the flat and stable pharmacokinetic profile of insulin degludec, as also shown previously [23], is preserved in subjects with impaired renal function, and that the exposure level of insulin degludec in a once-daily dosing regimen is comparable between subjects with renal impairment and subjects with normal renal function.
5 Conclusion
In conclusion, the results of this study indicate that the ultra-long pharmacokinetic properties of insulin degludec are preserved in patients with renal impairment. While insulin dose must always be adjusted on an individual basis, this trial suggests that the pharmacokinetic properties of insulin degludec are not significantly influenced by renal function, and thus specific dose adjustment may not be required for subjects with renal impairment. Accordingly, insulin degludec represents a useful insulin treatment option in subjects with co-morbid diabetes and impaired renal function, including ESRD patients undergoing hemodialysis.
References
Cavanaugh KL. Diabetes management issues for patients with chronic kidney disease. Clinical Diabetes. 2007;25:90–7.
de Boer IH, Rue TC, Hall YN, et al. Temporal trends in the prevalence of diabetic kidney disease in the United States. JAMA. 2001;305:2532–9.
Middleton RJ, Foley RN, Hegarty J, et al. The unrecognized prevalence of chronic kidney disease in diabetes. Nephrol Dial Transplant. 2006;21:88–92.
Assogba GF, Couchoud C, Roudier C, et al. Prevalence, screening and treatment of chronic kidney disease in people with type 2 diabetes in France: the ENTRED surveys (2001 and 2007). Diabetes Metab. 2012;38:558–66.
New JP, Middleton RJ, Klebe B, et al. Assessing the prevalence, monitoring and management of chronic kidney disease in patients with diabetes compared with those without diabetes in general practice. Diabet Med. 2007;24:364–9.
Harvey JN. Trends in the prevalence of diabetic nephropathy in type 1 and type 2 diabetes. Curr Opin Nephrol Hypertens. 2003;12:317–22.
Iglesias P, Diez JJ. Insulin therapy in renal disease. Diabetes Obes Metab. 2008;10:811–23.
Biesenbach G, Raml A, Schmekal B, Eichbauer-Sturm G. Decreased insulin requirement in relation to GFR in nephropathic type 1 and insulin-treated type 2 diabetic patients. Diabet Med. 2003;20:642–5.
Moen MF, Zhan M, Hsu VD, et al. Frequency of hypoglycemia and its significance in chronic kidney disease. Clin J Am Soc Nephrol. 2009;2009(4):1121–7.
National Kidney Foundation KDOQI. KDOQI clinical practice guidelines for diabetes and CKD: 2012 update; 2012. Available from: http://www.kidney.org/professionals/KDOQI/guidelines_diabetesUp/diabetes-ckd-update-2012.pdf. Accessed 28 Jun 2013
Muhlhauser I, Toth G, Sawicki PT, Berger M. Severe hypoglycemia in type I diabetic patients with impaired kidney function. Diabetes Care. 1991;14:344–6.
KDOQI. KDOQI clinical practice guidelines and clinical practice recommendations for diabetes and chronic kidney disease; 2007. Available from: http://www.kidney.org/professionals/kdoqi/pdf/Diabetes_AJKD_FebSuppl_07.pdf. Accessed 28 Jun 2013
Farrokhi F, Klindukhova O, Chandra P, et al. Risk factors for inpatient hypoglycemia during subcutaneous insulin therapy in non-critically ill patients with type 2 diabetes. J Diabetes Sci Technol. 2012;6:1022–9.
Scheen AJ. Pharmacokinetic considerations for the treatment of diabetes in patients with chronic kidney disease. Expert Opin Drug Metab Toxicol. 2013;9:529–50.
Snyder RW, Berns JS. Use of insulin and oral hypoglycemic medications in patients with diabetes mellitus and advanced kidney disease. Semin Dial. 2004;17:365–70.
Rave K, Heise T, Pfützner A, et al. Impact of diabetic nephropathy on pharmacodynamic and pharmacokinetic properties of insulin in type 1 diabetic patients. Diabetes Care. 2001;24:886–90.
Holmes G, Galitz L, Hu P, Lyness W. Pharmacokinetics of insulin aspart in obesity, renal impairment, or hepatic impairment. Br J Clin Pharmacol. 2005;60:469–76.
Lynes W, Tyler JF, Lawrence A. Renal impairment does not affect insulin aspart pharmacokinetics in type 1 diabetes. Diabetes. 2001;50(suppl 2):A441–2.
Novo Nordisk A/S. Levemir® EPAR. European Medicines Agency; 2012. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000528/WC500036662.pdf. Accessed 28 Jun 2013
Jacobsen LV, Popescu G, Plum A. Pharmacokinetics of insulin detemir in subjects with renal or hepatic impairment. Diabetologia. 2002;45(suppl 1):A259.
Kurtzhals P, Heise T, Strauss HM, et al. Multi-hexamer formation is the underlying basis for the ultra-long glucose-lowering effect of insulin degludec. Diabetologia. 2011;54(Suppl 1):S426.
Jonassen I, Havelund S, Hoeg-Jensen T, et al. Design of the novel protraction mechanism of insulin degludec, an ultra-long-acting basal insulin. Pharm Res. 2012;29:2104–14.
Heise T, Nosek L, Bøttcher SG, et al. Ultra-long-acting insulin degludec has a flat and stable glucose-lowering effect in type 2 diabetes. Diabetes Obes Metab. 2012;14:944–50.
Heise T, Hermanski L, Nosek L, et al. Insulin degludec: four times lower pharmacodynamic variability than insulin glargine under steady-state conditions in type 1 diabetes. Diabetes Obes Metab. 2012;14:859–64.
Birkeland KI, Home PD, Wendisch U, et al. Insulin degludec in type 1 diabetes: a randomized controlled trial of a new-generation ultra-long-acting insulin compared with insulin glargine. Diabetes Care. 2011;34:661–5.
Heller S, Buse J, Fisher M, et al. Insulin degludec, an ultra-long-acting basal insulin, versus insulin glargine in basal-bolus treatment with mealtime insulin aspart in type 1 diabetes (BEGIN Basal-Bolus Type 1): a phase 3, randomised, open-label, treat-to-target non-inferiority trial. Lancet. 2012;379:1489–97.
Garber AJ, King AB, Del Prato S, et al. Insulin degludec, an ultra-longacting basal insulin, versus insulin glargine in basal-bolus treatment with mealtime insulin aspart in type 2 diabetes (BEGIN Basal-Bolus Type 2): a phase 3, randomised, open-label, treat-to-target non-inferiority trial. Lancet. 2012;379:1498–507.
Ratner RE, Gough SC, Mathieu C, et al. Hypoglycaemia risk with insulin degludec compared with insulin glargine in type 2 and type 1 diabetes: a pre-planned meta-analysis of phase 3 trials. Diabetes Obes Metab. 2013;15:175–84.
Committee for Medicinal Products for Human Use. Note for guidance on the evaluation of the pharmacokinetics of medicinal products in patients with impaired renal function; 2004. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003123.pdf. Accessed 28 Jun 2013
US Department of Health and Human Services, Food and Drug Administration. Guidance for industry. Pharmacokinetics in patients with impaired renal function: study design, data analysis, and impact on dosing and labeling; 1998. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm072127.pdf. Accessed 28 Jun 2013
American Diabetes Association. Defining and reporting hypoglycemia in diabetes: a report from the American Diabetes Association Workgroup on Hypoglycemia. Diabetes Care. 2005;28:1245–9.
Novo Nordisk. Insulin degludec (NDA 203314) and insulin degludec/insulin aspart (NDA 203313) treatment to improve glycemic control. Briefing document for Endocrinologic and Metabolic Drug Advisory Committee; 2012. Available from: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/EndocrinologicandMetabolicDrugsAdvisoryCommittee/UCM327017.pdf. Accessed 28 Jun 2013
Baldwin D, Zander J, Munoz C, et al. A randomized trial of two weight-based doses of insulin glargine and glulisine in hospitalized subjects with type 2 diabetes and renal insufficiency. Diabetes Care. 2012;35:1970–4.
Pscherer S, Schreyer-Zell G, Gottsmann M. Experience with insulin glargine in patients with end-stage renal disease. Diabetes. 2002;216-OR:A53.
Sanofi. Lantus® EPAR. European Medicines Agency; 2012. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000284/WC500036082.pdf. Accessed 28 Jun 2013
Sanofi Aventis. Insuman® Comb. Summary of product characteristics; 2012. Available from: http://www.medicines.org.uk/emc/medicine/26470/SPC/Insuman+Comb+15+100+IU+ml+suspension+for+injection+in+a+cartridge/. Accessed 28 Jun 2013
Eli Lilly and Company. Humulin®. Summary of product characteristics; 2012. Available from: http://www.medicines.org.uk/emc/medicine/3425/SPC/Humulin+Vials%2c+Cartridges+and+KwikPens/. Accessed 28 Jun 2013
Novo Nordisk A/S. Insulatard®. Summary of product characteristics; 2012. Available from: http://www.medicines.org.uk/emc/medicine/3512/SPC/Insulatard+100+IU+ml%2c+Insulatard+Penfill+100+IU+ml%2c+Insulatard+InnoLet+100+IU+ml/. Accessed 28 Jun 2013
Novo Nordisk A/S. Tresiba®. Summary of product characteristics; 2013. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002498/WC500138940.pdf. Accessed 4 Oct 2013
Acknowledgments
The involvement of the St Imre Teaching Hospital in Budapest is gratefully acknowledged. Medical writing assistance was provided by ApotheCom ScopeMedical, UK, funded by Novo Nordisk. This study was funded by Novo Nordisk.
Disclosures
István Kiss has served on advisory panels for Abbot, Amgen, MSD and Novartis-Sandoz, and has served as a consultant to EGIS and Sanofi-Aventis. Carsten Roepstorff and Hanne Haahr are employees of Novo Nordisk A/S and are shareholders. Susanne Gammelgaard Bøttcher and Søren Klim are employees of Novo Nordisk A/S. Gerhard Arold has no disclosures.
Author contributions
István Kiss contributed to data interpretation, drafting and revising the manuscript; Gerhard Arold contributed to research design and execution, and revising the manuscript; Carsten Roepstorff contributed to data interpretation, drafting and revising the manuscript; Susanne Gammelgaard Bøttcher contributed to data analysis and revising the manuscript; Søren Klim contributed to data analysis and revised the manuscript; Hanne Haahr contributed to research design, data interpretation, drafting the manuscript and revising the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Kiss, I., Arold, G., Roepstorff, C. et al. Insulin Degludec: Pharmacokinetics in Patients with Renal Impairment. Clin Pharmacokinet 53, 175–183 (2014). https://doi.org/10.1007/s40262-013-0113-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-013-0113-2