
Abstract
Aims/hypothesis
MODY can be wrongly diagnosed as type 1 diabetes in children. We aimed to find the prevalence of MODY in a nationwide population-based registry of childhood diabetes.
Methods
Using next-generation sequencing, we screened the HNF1A, HNF4A, HNF1B, GCK and INS genes in all 469 children (12.1%) negative for both GAD and IA-2 autoantibodies and 469 antibody-positive matched controls selected from the Norwegian Childhood Diabetes Registry (3882 children). Variants were classified using clinical diagnostic criteria for pathogenicity ranging from class 1 (neutral) to class 5 (pathogenic).
Results
We identified 58 rare exonic and splice variants in cases and controls. Among antibody-negative patients, 6.5% had genetic variants of classes 3–5 (vs 2.4% in controls; p = 0.002). For the stricter classification (classes 4 and 5), the corresponding number was 4.1% (vs 0.2% in controls; p = 1.6 × 10−5). HNF1A showed the strongest enrichment of class 3–5 variants, with 3.9% among antibody-negative patients (vs 0.4% in controls; p = 0.0002). Antibody-negative carriers of variants in class 3 had a similar phenotype to those carrying variants in classes 4 and 5.
Conclusions/interpretation
This is the first study screening for MODY in all antibody-negative children in a nationwide population-based registry. Our results suggest that the prevalence of MODY in antibody-negative childhood diabetes may reach 6.5%. One-third of these MODY cases had not been recognised by clinicians. Since a precise diagnosis is important for treatment and genetic counselling, molecular screening of all antibody-negative children should be considered in routine diagnostics.
Similar content being viewed by others


Introduction
Monogenic forms of diabetes are caused by mutations in single genes and constitute a yet-unknown fraction of all diabetes. MODY is the most common type of monogenic diabetes and can be caused by mutations in at least 13 genes [1]. The clinical picture is characterised by autosomal dominant inheritance, onset before 25–35 years of age, normal body weight, primary beta cell dysfunction and lack of markers of autoimmune diabetes. In the Norwegian MODY Registry, the distribution of the most common MODY forms among patients with a molecular diagnosis of MODY is as follows: HNF1A-MODY 53%, GCK-MODY 30%, HNF4A-MODY 7.5% and HNF1B-MODY 5.6%, while several other forms are very rare [2–4].
Clinically, MODY may be difficult to distinguish from type 1 and type 2 diabetes. It is therefore likely that many patients with monogenic forms are misclassified. A molecular diagnosis of monogenic diabetes is essential for optimal treatment, prognosis and genetic counselling. Patients with MODY caused by a mutation in the HNF1A or HNF4A genes are sensitive to sulfonylurea treatment, which is associated with improved glycaemic control and quality of life [5–9]. Moreover, patients with a mutation in GCK typically do not require pharmacological intervention [10]. Hence, individuals with unrecognised MODY may receive suboptimal treatment.
Although we and others have estimated the prevalence of monogenic diabetes to be 1.1–4.2% of all children with diabetes [11–14], no study has yet screened a nationwide population-based cohort in a systematic way.
Various guidelines have been established to aid clinicians in recognising patients who might have clinical signs of MODY, such as lack of diabetes autoantibodies, normal BMI, sustained insulin secretion capacity years after diagnosis and a family history of diabetes. Studies indicate that lower levels of the biomarker high-sensitivity C-reactive protein (hs-CRP) can predict HNF1A-MODY patients for diagnostic genetic testing [15].
Despite this, a substantial fraction of patients with MODY remain undiagnosed. Thus, current guidelines, combined with clinical ‘intuition’, are imperfect compared with molecular genetic testing. To circumvent these challenges, we designed our study to screen the MODY genes HNF1A, HNF4A, HNF1B, GCK and INS by next-generation sequencing in all antibody-negative children and controls from the population-based Norwegian Childhood Diabetes Registry (NCDR).
Methods
Study design
We selected all 469 children in the NCDR who were negative for both GAD and IA-2 autoantibodies at diabetes onset. The control group was also selected from the NCDR and consisted of 469 age- and sex-matched children positive for both GAD and IA-2.
Study population
All children less than 15 years of age developing diabetes (using WHO criteria) in Norway are invited to participate in the NCDR. Estimates show that the NCDR contains data from more than 96% of all children diagnosed with diabetes since 2002 [16]. By March 2015, the registry contained clinical data (onset of diabetes, family history, treatment, BMI, etc.) as well as serum and DNA samples for 3882 children. In Norway, GAD and IA-2 autoantibodies are analysed in all children developing diabetes and these data are supplied to the NCDR.
Laboratory analyses
Autoantibodies to GAD and IA-2 were analysed at Haukeland University Hospital or Oslo University Hospital, Aker using a radioligand assay in which recombinant anti-GAD and anti-IA-2 were labelled and detected as described [12]. The cut-off criteria for autoantibody negativity were antibody index < 0.08 or concentration < 1.0 U/ml for GAD, and antibody index < 0.1 or concentration < 1.0 U/ml for IA-2.
Genetic analyses
We extracted genomic DNA (gDNA) from peripheral leucocytes using standard procedures.
The selected panel, TruSeq custom amplicon (Illumina, Eindhoven, the Netherlands), covers a total of 62,525 bp. The 425 bp amplicons were designed to cover the entire coding region and exon–intron boundaries (±5 bp). In addition, 2 kb of the flanking sequence in the 5′ UTR and some specific regions, promoter regions in HNF4A (c.-192), GCK (c.-557) and INS intron (c.188-31), were added to the target regions of selected genes. Adaptor-ligated libraries were prepared from 250 ng of gDNA following protocol instructions (Illumina). Amplicons of 938 blood samples were sequenced on an Illumina MiSeq sequencer at Hudson Alpha Institute for Biotechnology (Huntsville, AL, USA). The sequencing of 250 bp pair-end reads resulted in an average of 82 Mbp of raw sequence data per sample. Six samples were excluded due to low coverage. The panel set-up included a total of 16 genes, although in this study we investigated the 13 MODY genes: HNF1A, GCK, HNF4A, HNF1B, INS, ABCC8, KCNJ11, BLK, CEL, NEUROD1, KLF11, PAX4 and PDX1. The reference sequences used for alignment were: GCK, NM_000162.3; HNF1A, NM_000545.6; HNF1B, NM_000458.3; HNF4A, NM_175914.4; INS, NM_000207.2; ABCC8, NM_000352.3; BLK, NM_001715; CEL, NM_01807; KCNJ11, NM_000525.3; KLF11, NM_001177716.1; NEUROD1, NM_002500.3; PAX4, NM_006193.2 and PDX1, NM_000209.3. The genomic coordinates for the full gene panel are listed in the electronic supplementary material [ESM Table 1]. Alignments and variant calling were performed using TruSeq Amplicon workflow (v.1.0.0.57; GATK variant caller) in the Illumina BaseSpace analysis software (v.2.4.60.8). Low-quality variant calls (depth < 20, variant quality < 100) were removed from the variant lists. Next, to retain only rare variants, we excluded variants with minor allele frequency (MAF) > 1% in the 1000 Genomes database (phase 1 release v3 called from 20101123 alignment) [17], variants present with frequency ≥ 1% in an in-house database of over 300 Norwegian exomes and variants present among the patients with a frequency higher than 30%. Our in-house database of allele frequencies encompasses 300 exomes derived from research projects on a diverse set of rare non-diabetes-related Mendelian disorders. All analyses were performed on rare variants within coding sequences as defined by RefSeq (https://www.ncbi.nlm.nih.gov/refseq/), and both filtering and categorisation of variants were done using ANNOVAR [18]. Statistics on variant counts at different filtering stages, as well as detailed gene/exon coverage statistics, can be found in ESM Table 2 and ESM Fig. 1.
We used DNA samples from 22 patients with known variants in the five selected genes as controls. We confirmed all newly identified variants by Sanger sequencing.
Classification of sequence variants
All rare variants (MAF <1%) were classified using a five-tier score system as defined by Plon et al [19]. This classification model is commonly used in clinical diagnostics laboratories and places a variant into one of five groups: class 1, neutral/benign; class 2, likely neutral/benign; class 3, unknown significance; class 4, likely pathogenic and class 5, pathogenic. We scored the variants based on criteria using clinical, functional and segregation data from the literature (PubMed, HGMD professional [www.hgmd.cf.ac.uk/] and ClinVar [www.ncbi.nlm.nih.gov/clinvar/]) and population data (ExAC [http://exac.broadinstitute.org/], 1000 Genomes [www.ncbi.nlm.nih.gov/variation/tools/1000genomes/], ESP [http://evs.gs.washington.edu/EVS/] and dbSNP [www.ncbi.nlm.nih.gov/snp]), as well as information from the Norwegian MODY Registry. Splice site predictions were performed by the integrated software of Alamut v.2.6 (Interactive Biosoftware, Rouen, France) using default settings. The 1–5 classification system puts less emphasis on bioinformatics and the in silico predictions (SIFT, PolyPhen-2, Align GVGD, Mutation Taster and CADD) are used only as supportive information. It is also important to mention that this classification model is relevant and applicable to Mendelian disease (and not complex disorders). For a variant to be classified as class 5, clinical, functional and segregation data must be present. Nonsense, frameshift or essential splice site variations were, by their nature, considered pathogenic or likely pathogenic.
Classification of DNA variant pathogenicity was carried out independently by three researchers (BBJ, JM and IA) and performed blindly to autoantibody status.
Statistical analysis
We compared clinical observations between antibody-negative and antibody-positive individuals (Table 1). For antibody-negative individuals, we compared those harbouring variants in classes 3–5 with those harbouring class 1 and 2 variants. Dichotomous outcome was studied using binomial regression with log link to estimate RR with CI between groups. Comparisons of the mean of groups were conducted using the two-sample t test. BMI standard deviation scores (SDS) were calculated using the current references for Norwegian children [20]. All statistical analyses were performed using the STATA statistic Software (version 14.0) (StataCorp LP, College Station, TX, USA).
Ethics
The present study was conducted in accordance with the Declaration of Helsinki and approved by the Regional Committee for Research Ethics. Written informed consent was obtained from all participants or their guardians.
Results
Targeted deep sequencing
In this study, a panel of 13 MODY genes was screened (HNF1A, HNF4A, HNF1B, GCK, INS, ABCC8, KCNJ11, BLK, CEL, NEUROD1, KLF11, PAX4 and PDX1) in a cohort of 938 patients from the NCDR using TruSeq Custom Amplicon targeted sequencing. The mean depth of coverage across samples was between 207× and 1186.1×, with between 81.1% and 99.2% of the base pairs covered by at least 20 reads (ESM Table 2 and ESM Fig. 1). We mainly focused on the five MODY genes HNF1A, HNF4A, HNF1B, GCK and INS, which constitute about 98% of all MODY in Norway.
To validate our assay, 22 samples with known variants in the selected target genes previously confirmed by Sanger sequencing were run as controls. All these variants were identified correctly by the TruSeq custom panel. Variants detected in the screening described here were checked by Sanger sequencing and no false positives were detected.
As choice of annotation software might affect the results [21, 22], we tested both RefSeq and GENCODE with several types of annotation software. Results were largely consistent between software types and no additional variants in classes 3–5 were detected. Hence, the results are presented with strategy as described in Methods.
Frequency of MODY variants in HNF1A, HNF4A, HNF1B, GCK and INS in the study population
We analysed all children (n = 469) in the NCDR who were negative for both GAD and IA-2 autoantibodies. As controls, we selected 469 age- and sex-matched patients who were positive for both GAD and IA-2 autoantibodies. Baseline characteristics of the study participants are presented in Table 1.
We identified 58 individuals carrying rare exonic and putative splice variants in the five main target genes. No patient had more than one rare variant in the genes analysed.
Using bioinformatics tools, literature and the Norwegian MODY Registry (see Methods), we could classify 34 of the 58 variants to be of unknown significance, likely pathogenic or pathogenic (classes 3–5). Of these 34 variants, 22 have previously been reported and 12 are novel (Table 2). A total of 37 patients in the antibody-negative group had a rare variant (classes 2–5) as compared with 21 patients in the antibody-positive group. Seventeen individuals had variants that were considered likely benign (class 2), whereas 41 patients had variants ranging from unknown significance to pathogenic (classes 3–5) (Fig. 1). Hence, 6.5% (30/462) of the autoantibody-negative children had variants of class 3–5 compared with 2.4% (11/468) of the autoantibody-positive children (p = 0.002, RR 2.8 [95% CI 1.4, 5.5]) (Fig. 2). Interestingly, 3.9% of the autoantibody-negative patients had an HNF1A variant of class 3–5 compared with 0.4% of the autoantibody-positive patients (p = 0.0002, RR 9.1 [95% CI 2.2, 39.1]). The distribution of the different variants throughout the HNF1A gene is shown in Fig. 3.

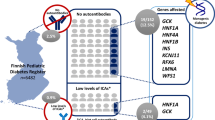
Outline of the study. Antibody-negative and -positive individuals are defined, respectively, as individuals negative or positive for both GAD and IA-2. Rare variants are in HNF1A, HNF4A, HNF1B, GCK and INS with MAF <1% in 1000 Genomes, in an in-house database of 300 Norwegian exomes with frequency <1%, and in the patients with a frequency <30%

Distribution of variants (%) in pathogenicity classes 3–5 in antibody-negative and antibody-positive patients in the NCDR. Pink bars, antibody-negative class 3; red bars, antibody-negative class 4/5; light blue bars, antibody-positive class 3; dark blue bars, antibody-positive class 4/5. **p = 0.002 and ***p = 0.0002

Schematic representation of HNF1A, the corresponding functional domains of the protein and locations of variants detected in the NCDR. Blue star denotes non-synonymous variant, antibody-positive; red star denotes non-synonymous variant, antibody-negative; red circle denotes insertion/deletion, antibody-negative
Using a stricter measure for pathogenicity, 4.1% (19/462) of the antibody-negative patients and 0.2% (1/468) of the antibody-positive patients carried a pathogenic variant of class 4 or 5 (p = 1.6 × 10−5, RR 19.3 [95% CI 2.6, 143.2]).
In the Norwegian MODY Registry, 21 of the 30 patients harbouring variants in classes 3–5 had been identified previously by clinical criteria alone, reflecting a high level of awareness for MODY in Norway. However, we still detected nine new individuals with MODY in this study. Five of these nine individuals who had not been referred for MODY testing carried variants evaluated as class 3 and four individuals carried class 4 or class 5 variants. NCDR data revealed that the latter four individuals had no known family history, indicating the presence of de novo variants and/or that family history had been under-reported in clinical records. Unfortunately, the parents were unavailable for testing.
Of the 11 variants detected in the antibody-positive group, all but one were scored as class 3. The single class 4 variant was identified in GCK. It seems, however, that this child had double diabetes (i.e. GCK-MODY and type 1 diabetes), since he/she at diagnosis had no detectable levels of C-peptide, no family history of insulin-dependent diabetes, age of onset at 2.1 years, an HbA1c at 13.1% (120 mmol/mol) and insulin requirement. The clinical characteristics of all children in the NCDR identified in this study as harbouring a genetic variant of classes 3–5 are listed in Table 3.
Clinical characteristics of the carriers of class 3–5 variants
Linking genetic information to clinical characteristics of patients from the NCDR (Table 4) revealed that age of onset was significantly higher for the carriers of class 3–5 variants compared with non-carriers in the antibody-negative group (11.1 vs 9.0 years; p = 0.004). The probability of having a family history of diabetes, HbA1c < 7.5% (<58 mmol/mol) or being treated by diet or a sulfonylurea were significantly higher for this group. Treatment with insulin was less common among the autoantibody-negative carriers of class 3–5 variants compared with the non-carriers (p < 0.001). Data from the last annual check-up showed that antibody-negative patients with a class 3–5 variant had lower HbA1c (p = 0.058), lower daily insulin consumption (p < 0.001) and higher BMI SDS (p = 0.004) when compared with the individuals without a variant in the five analysed genes.
Our data allow us to address whether patients with variants of uncertain significance (class 3) were similar, clinically, either to carriers of class 4 or 5 variants (MODY), indicating that these variants are likely pathogenic, or to patients without variants (type 1 diabetes, not MODY). We therefore investigated both antibody-negative and antibody-positive children and compared the clinical characteristics of the three different groups. Patients with class 3 variants in the autoantibody-negative group were not significantly different from the MODY variant (classes 4 and 5) carriers but were different from the antibody-negative patients without class 3–5 variants (ESM Tables 3 and 4). However, for the autoantibody-positive patients, the variant carriers (class 3 and 4) showed no significant difference in clinical characteristics when compared with the individuals without class 3 or 4 variants, but the numbers studied were small.
Discussion
To our knowledge, this is the first study to investigate the prevalence of MODY in all antibody-negative children with diabetes in a national diabetes registry by sequencing the five most common MODY genes. A total of 6.5% of these children had variants in pathogenicity class 3–5, while 4.1% had variants in class 4 or 5. Importantly, when evaluating clinical data, we observed that carriers of class 3 variants had clinical features typical of MODY (family history, HbA1c, insulin use, sulfonylurea treatment and age of onset), similar to patients selected by the stricter classification (classes 4 and 5) (Table 4). Thus, we believe that most patients with class 3 variants in the antibody-negative group are likely to have MODY. In the control group, positive for anti-GAD and anti-IA-2, we found that 2.4% had a variant of class 3–5 (n = 10 scored as class 3 and only 1 as class 4). However, the clinical characteristics of these patients were compatible with type 1 diabetes (ESM Table 4). Thus, it is likely that these variants represent the background population frequency of very rare benign variants, which are difficult to classify due to lack of information. Indeed, as many as 10 of 11 of these variants were scored as variants of unknown significance by all three evaluators who were blinded to case/control status.
We also screened for variants in seven other rare causes of MODY genes (ABCC8, KCNJ11, BLK, KLF11, NEUROD1, PAX4 and PDX1) (presented in ESM Tables 5 and 6). CEL was not included due to poor sequence quality. Sequence variants identified in ABCC8 and KCNJ11, that can cause both permanent neonatal diabetes, hyperinsulinaemic hypoglycaemia and probably MODY, were classified with respect to MODY (ESM Table 5). The four class 4 and 5 ABCC8 variants associated with congenital hyperinsulinism were excluded from the statistical analysis. Of the remaining 17 individuals with a class 3–5 variant in ABCC8 or KCNJ11, one patient diagnosed with diabetes at 2 months of age had the recurrent p.Val59Met class 5 KCNJ11 variant known to cause permanent neonatal diabetes [23]. This patient was known to have KCNJ11-permanent neonatal diabetes and was treated with a sulfonylurea. All the other variants were classified as being of unknown significance (class 3), due to there being little or no available evidence to support or refute their possible pathogenicity. Notably, there was no difference in the percentage of class 3–5 variant carriers for these two genes when comparing antibody-negative patients with antibody-positive patients, suggesting that most are benign. Furthermore, the clinical characteristics of the antibody-negative carriers of class 3–5 variants in ABCC8 and KCNJ11 showed no differences from those without variants. An exception is the class 5 variant in KCNJ11 causing permanent neonatal diabetes (ESM Table 7).
We detected 52 rare variants in the remaining five MODY genes (BLK, KLF11, NEUROD1, PAX4 and PDX1). We could not classify these variants due to lack of published information regarding their clinical significance; only the bioinformatics interpretation is presented (ESM Table 6). We found no differences in frequencies between antibody-negative and antibody-positive patients.
Our main finding that at least up to 6.5% of antibody-negative children with diabetes have MODY is somewhat lower than the proportion reported in other studies [11–14]. Still, our data might be more robust. The NCDR includes close to all children less than 15 years of age with newly diagnosed diabetes in Norway. Thus, our data should give a precise number of the true prevalence of MODY in the most relevant population of children with diabetes (i.e. the antibody-negative cases). It should be noted that the NCDR holds data from children with newly discovered diabetes and who are admitted to hospitals that have robust routines for inclusion in the registry. Thus, cases diagnosed in outpatient clinics are likely under-represented. Hence, it is not surprising that the frequency of GCK mutations found in our study is much lower than in a recent study performed in clinics with particular MODY awareness [14]. Additionally, the age limit of most children’s hospitals in Norway is 15 years of age, which is likely in the lower range of age of diagnosis of MODY. A somewhat higher prevalence may be found in countries that use a higher age limit for defining paediatric diabetes.
A precise diagnosis of MODY is important for treatment and genetic counselling. Patients with pathogenic GCK variants usually need no pharmacological treatment, except during pregnancy when close monitoring is needed since the relative insulin resistance often leads to more severe diabetes with a need for insulin [24]. It should, however, be noted that some patients might have double diabetes (i.e. both type 1 diabetes and GCK-MODY). For individuals carrying pathogenic HNF1A or HNF4A variants, sulfonylurea is the treatment of choice. Table 3 illustrates 5 of 19 individuals in the antibody-negative group who were carriers of pathogenic HNF1A or HNF4A variants and used insulin when discharged from hospital 2 weeks after diabetes diagnosis. This indicates that the clinical presentation of these forms of MODY might be difficult to distinguish from that of type 1 diabetes at diagnosis. A precise molecular diagnosis, if known, therefore may lead to change of treatment. Thus, this study may lead to a change in treatment in patients carrying pathogenic variants. For the novel variants discovered, we will perform functional testing that will be included in the evaluation of possible change of treatment [25]. The high risk of recurrence for MODY is important regarding genetic counselling.
We found that clinical criteria alone are not sufficient to detect all MODY patients in a population, as around one-third of the individuals with MODY in the Norwegian paediatric diabetes population had not been referred to molecular screening using current clinical practise. Reasons for this might include lack of awareness about the MODY phenotype and criteria for genetic testing in children with diabetes. Thus, there is a need for better information and more robust ‘best practise’ for diagnosing MODY. It might now be time to consider screening for the common MODY genes in all children and adolescents who develop autoantibody-negative diabetes.
Prior to this study, two-thirds of the patients carrying MODY variants were already diagnosed, most likely explaining why individuals carrying a class 3–5 variant in one of the five MODY genes had lower HbA1c levels and insulin requirements than individuals with a class 1 or 2 variant. HbA1c levels were similar in patients harbouring a class 3 variant and those harbouring class 4 or 5 variants but not class 1 or 2 variants. This is interesting since several in the class 3 variant group had not received a MODY diagnosis prior to this study. Thus, individuals with class 3 variants, as well as those with class 4 or 5 variants, respond to personalised treatment, with improved metabolic control. It is therefore important to identify individuals who carry class 3–5 variants and ensure that they receive correct treatment. Hence, we believe a precise diagnosis will improve treatment of patients with impaired fasting glucose and diabetic patients, provide more appropriate genetic counselling and enable better prediction of diabetes-related late complications.
There are limitations to our study. We did not screen all the 3882 children with type 1 diabetes in the NCDR. Genetic screening of children who were positive for only one of the two autoantibodies might increase the diagnostic yield [26]. We did not include antibodies against insulin and zinc transporter 8 (ZnT8) in our study since these were not part of routine antibody screening when the NCDR started in 2002. Inclusion of ZnT8 antibodies are now included in our routine screen. Also, the upper normal limit for anti-GAD and anti-IA-2 is ≤ 1 U/ml and setting the cut-off at a lower level might refine the results even more.
Recently, a new biomarker for HNF1A, high-sensitivity C-reactive protein, has been reported [15, 27]. Presently, this biomarker is not in routine use in our registry. Urinary and serum C-peptide levels persist in patients with MODY who retain endogenous beta cell function [28, 29]. A recent study from the UK showed that the biomarkers efficiently identify paediatric patients appropriate for genetic screening [14]. These markers have not been included in our routine screening since the analysis in Norway is done when diabetes is detected. At the time of diagnosis some children with type 1 diabetes will have an endogenous insulin production and the test would therefore not be able to differentiate between the groups. A new test after some years would be more valuable, but has probably not been included in the follow-up programme in Norway since antibodies are measured at diagnosis.
In conclusion, we found that 4.1–6.5% of children with antibody-negative diabetes, listed in the NCDR, have sequence variants that most likely are pathogenic and compatible with a diagnosis of MODY. Moreover, one-third of the cases were not picked up by clinical criteria alone. We therefore suggest screening all antibody-negative children for pathogenic variants in the most common MODY genes, to improve diagnosis and treatment as part of the new precision medicine.
Abbreviations
- IA-2:
-
Insulinoma-associated antigen 2
- MAF:
-
Minor allele frequency
- NCDR:
-
Norwegian Childhood Diabetes Registry
- SDS:
-
SD scores
References
Rubio-Cabezas O, Hattersley AT, Njolstad PR et al (2014) ISPAD clinical practice consensus guidelines 2014. The diagnosis and management of monogenic diabetes in children and adolescents. Pediatr Diabetes 15(suppl 20):47–64
Molven A, Njolstad PR (2011) Role of molecular genetics in transforming diagnosis of diabetes mellitus. Expert Rev Mol Diagn 11:313–320
Søvik O, Irgens HU, Molnes J et al (2013) Monogenic diabetes mellitus in Norway. Norsk Epidemiol 2013:55–60
Vaxillaire M, Bonnefond A, Froguel P (2012) The lessons of early-onset monogenic diabetes for the understanding of diabetes pathogenesis. Best Pract Res Clin Endocrinol Metab 26:171–187
Pearson ER, Liddell WG, Shepherd M, Corrall RJ, Hattersley AT (2000) Sensitivity to sulphonylureas in patients with hepatocyte nuclear factor-1α gene mutations: evidence for pharmacogenetics in diabetes. Diabet Med 17:543–545
Pearson ER, Starkey BJ, Powell RJ, Gribble FM, Clark PM, Hattersley AT (2003) Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet 362:1275–1281
Shepherd M, Hattersley AT (2004) ʻI donʼt feel like a diabetic any moreʼ: the impact of stopping insulin in patients with maturity onset diabetes of the young following genetic testing. Clin Med 4:144–147
Shepherd M, Shields B, Ellard S, Rubio-Cabezas O, Hattersley AT (2009) A genetic diagnosis of HNF1A diabetes alters treatment and improves glycaemic control in the majority of insulin-treated patients. Diabet Med 26:437–441
Thanabalasingham G, Pal A, Selwood MP et al (2012) Systematic assessment of etiology in adults with a clinical diagnosis of young-onset type 2 diabetes is a successful strategy for identifying maturity-onset diabetes of the young. Diabetes Care 35:1206–1212
Ajjan RA, Owen KR (2014) Glucokinase MODY and implications for treatment goals of common forms of diabetes. Curr Diab Rep 14:559
Fendler W, Borowiec M, Baranowska-Jazwiecka A et al (2012) Prevalence of monogenic diabetes amongst Polish children after a nationwide genetic screening campaign. Diabetologia 55:2631–2635
Irgens HU, Molnes J, Johansson BB et al (2013) Prevalence of monogenic diabetes in the population-based Norwegian Childhood Diabetes Registry. Diabetologia 56:1512–1519
Pihoker C, Gilliam LK, Ellard S et al (2013) Prevalence, characteristics and clinical diagnosis of maturity onset diabetes of the young due to mutations in HNF1A, HNF4A, and glucokinase: results from the SEARCH for Diabetes in Youth. J Clin Endocrinol Metab 98:4055–4062
Shepherd M, Shields B, Hammersley S et al (2016) Systematic population screening, using biomarkers and genetic testing, identifies 2.5% of the U.K. Pediatric Diabetes Population With Monogenic Diabetes. Diabetes Care 39:1879–1888
Thanabalasingham G, Shah N, Vaxillaire M et al (2011) A large multi-centre European study validates high-sensitivity C-reactive protein (hsCRP) as a clinical biomarker for the diagnosis of diabetes subtypes. Diabetologia 54:2801–2810
Skrivarhaug T, Stene LC, Drivvoll AK, Strom H, Joner G, Norwegian Childhood Diabetes Study G (2014) Incidence of type 1 diabetes in Norway among children aged 0-14 years between 1989 and 2012: has the incidence stopped rising? Results from the Norwegian Childhood Diabetes Registry. Diabetologia 57:57–62
Genomes Project C, Abecasis GR, Auton A et al (2012) An integrated map of genetic variation from 1,092 human genomes. Nature 491:56–65
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38, e164
Plon SE, Eccles DM, Easton D et al (2008) Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat 29:1282–1291
Juliusson PB, Roelants M, Nordal E et al (2013) Growth references for 0-19 year-old Norwegian children for length/height, weight, body mass index and head circumference. Ann Hum Biol 40:220–227
Frankish A, Uszczynska B, Ritchie GR et al (2015) Comparison of GENCODE and RefSeq gene annotation and the impact of reference geneset on variant effect prediction. BMC Genomics 8(16 Suppl):S2
McCarthy DJ, Humburg P, Kanapin A et al (2014) Choice of transcripts and software has a large effect on variant annotation. Genome Med 6:26
Sagen JV, Raeder H, Hathout E et al (2004) Permanent neonatal diabetes due to mutations in KCNJ11 encoding Kir6.2: patient characteristics and initial response to sulfonylurea therapy. Diabetes 53:2713–2718
Stride A, Shields B, Gill-Carey O et al (2014) Cross-sectional and longitudinal studies suggest pharmacological treatment used in patients with glucokinase mutations does not alter glycaemia. Diabetologia 57:54–56
Consortium STD, Estrada K, Aukrust I et al (2014) Association of a low-frequency variant in HNF1A with type 2 diabetes in a Latino population. JAMA 311:2305–2314
McDonald TJ, Colclough K, Brown R et al (2011) Islet autoantibodies can discriminate maturity-onset diabetes of the young (MODY) from type 1 diabetes. Diabet Med 28:1028–1033
McDonald TJ, Shields BM, Lawry J et al (2011) High-sensitivity CRP discriminates HNF1A-MODY from other subtypes of diabetes. Diabetes Care 34:1860–1862
Besser RE, Shepherd MH, McDonald TJ et al (2011) Urinary C-peptide creatinine ratio is a practical outpatient tool for identifying hepatocyte nuclear factor 1α/hepatocyte nuclear factor 4α maturity-onset diabetes of the young from long-duration type 1 diabetes. Diabetes Care 34:286–291
Besser RE, Shields BM, Hammersley SE et al (2013) Home urine C-peptide creatinine ratio (UCPCR) testing can identify type 2 and MODY in pediatric diabetes. Pediatr Diabetes 14:181–188
Sagen JV, Bjorkhaug L, Molnes J et al (2008) Diagnostic screening of MODY2/GCK mutations in the Norwegian MODY Registry. Pediatr Diabetes 9:442–449
Sagen JV, Odili S, Bjorkhaug L et al (2006) From clinicogenetic studies of maturity-onset diabetes of the young to unraveling complex mechanisms of glucokinase regulation. Diabetes 55:1713–1722
Njolstad PR, Sovik O, Cuesta-Munoz A et al (2001) Neonatal diabetes mellitus due to complete glucokinase deficiency. N Engl J Med 344:1588–1592
Beer NL, Osbak KK, van de Bunt M et al (2012) Insights into the pathogenicity of rare missense GCK variants from the identification and functional characterization of compound heterozygous and double mutations inherited in cis. Diabetes Care 35:1482–1484
Gidh-Jain M, Takeda J, Xu LZ et al (1993) Glucokinase mutations associated with non-insulin-dependent (type 2) diabetes mellitus have decreased enzymatic activity: implications for structure/function relationships. Proc Natl Acad Sci U S A 90:1932–1936
Molnes J, Bjorkhaug L, Sovik O, Njolstad PR, Flatmark T (2008) Catalytic activation of human glucokinase by substrate binding: residue contacts involved in the binding of D-glucose to the super-open form and conformational transitions. FEBS J 275:2467–2481
Garcia-Herrero CM, Rubio-Cabezas O, Azriel S et al (2012) Functional characterization of MODY2 mutations highlights the importance of the fine-tuning of glucokinase and its role in glucose sensing. PLoS One 7, e30518
Osbak KK, Colclough K, Saint-Martin C et al (2009) Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum Mutat 30:1512–1526
Bjorkhaug L, Ye H, Horikawa Y, Sovik O, Molven A, Njolstad PR (2000) MODY associated with two novel hepatocyte nuclear factor-1α loss-of-function mutations (P112L and Q466X). Biochem Biophys Res Commun 279:792–798
Glucksmann MA, Lehto M, Tayber O et al (1997) Novel mutations and a mutational hotspot in the MODY3 gene. Diabetes 46:1081–1086
Flannick J, Beer NL, Bick AG et al (2013) Assessing the phenotypic effects in the general population of rare variants in genes for a dominant Mendelian form of diabetes. Nat Genet 45:1380–1385
Ng MC, Cockburn BN, Lindner TH et al (1999) Molecular genetics of diabetes mellitus in Chinese subjects: identification of mutations in glucokinase and hepatocyte nuclear factor-1α genes in patients with early-onset type 2 diabetes mellitus/MODY. Diabet Med 16:956–963
Kaisaki PJ, Menzel S, Lindner T et al (1997) Mutations in the hepatocyte nuclear factor-1α gene in MODY and early-onset NIDDM: evidence for a mutational hotspot in exon 4. Diabetes 46:528–535
Thomas H, Badenberg B, Bulman M et al (2002) Evidence for haploinsufficiency of the human HNF1α gene revealed by functional characterization of MODY3-associated mutations. Biol Chem 383:1691–1700
Bjorkhaug L, Sagen JV, Thorsby P, Sovik O, Molven A, Njolstad PR (2003) Hepatocyte nuclear factor-1α gene mutations and diabetes in Norway. J Clin Endocrinol Metab 88:920–931
Yamagata K, Yang Q, Yamamoto K et al (1998) Mutation P291fsinsC in the transcription factor hepatocyte nuclear factor-1α is dominant negative. Diabetes 47:1231–1235
Ellard S, Bulman MP, Frayling TM, Shepherd M, Hattersley AT (2000) Proposed mechanism for a novel insertion/deletion frameshift mutation (I414G415ATCG → CCA) in the hepatocyte nuclear factor 1 alpha (HNF-1α) gene which causes maturity-onset diabetes of the young (MODY). Hum Mutat 16:273
Colclough K, Bellanne-Chantelot C, Saint-Martin C, Flanagan SE, Ellard S (2013) Mutations in the genes encoding the transcription factors hepatocyte nuclear factor 1 alpha and 4 alpha in maturity-onset diabetes of the young and hyperinsulinemic hypoglycemia. Hum Mutat 34:669–685
Ferre S, Bongers EM, Sonneveld R et al (2013) Early development of hyperparathyroidism due to loss of PTH transcriptional repression in patients with HNF1β mutations? J Clin Endocrinol Metab 98:4089–4096
Molven A, Ringdal M, Nordbo AM et al (2008) Mutations in the insulin gene can cause MODY and autoantibody-negative type 1 diabetes. Diabetes 57:1131–1135
Acknowledgements
The authors thank M. Ringdal, L. Aasmul and L. Grevle (all K. G. Jebsen Center for Diabetes Research, Department of Clinical Science, University of Bergen, Bergen, Norway) for expert technical assistance, Å. Irgens (Department of Occupational Medicine, Haukeland University Hospital) for statistical guidance, all the members of the Norwegian Childhood Diabetes Study Group, the NCDR and the Norwegian MODY Registry for their contribution and finally the patients who contributed their data.
Some of the data in this study were presented as an abstract at the 51st EASD Annual Meeting in Stockholm in 2015.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Data availability
The data generated during the current study are available from the corresponding author on reasonable request.
Funding
Funding was received from the Stiftelsen Kristian Gerhard Jebsen, Norwegian Diabetes Association, Western Norway Regional Health Authority, Southern and Eastern Norway Regional Health Authority, University of Bergen, Haukeland University Hospital, Novo Nordisk Foundation, Research Council of Norway and European Research Council (ERC AdG).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
BBJ, HUI, SJ and PRN developed the study concept and supervised the study. OS and AM helped with the study design. BBJ, JM and SL carried out the experimental work. BBJ, HUI, JM, PS, SL, PRN and SJ interpreted the data. PBJ contributed to the statistical work. TS was responsible for collecting the data from the NCDR. GJ helped with data collection and IA assisted in interpreting the data. BBJ drafted the first manuscript. All authors revised the manuscript critically for important intellectual content and approved the final version. BBJ and PRN are the guarantors for the paper, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Additional information
Henrik U. Irgens, Janne Molnes and Paweł Sztromwasser contributed equally to the study.
Stefan Johansson and Pål R. Njølstad share joint senior authorship.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Table 1
(XLSX 23.3 kb)
ESM 2
(PDF 768 kb)
Rights and permissions
About this article

Cite this article
Johansson, B.B., Irgens, H.U., Molnes, J. et al. Targeted next-generation sequencing reveals MODY in up to 6.5% of antibody-negative diabetes cases listed in the Norwegian Childhood Diabetes Registry. Diabetologia 60, 625–635 (2017). https://doi.org/10.1007/s00125-016-4167-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-4167-1