
Abstract
Aims/hypothesis
Monogenic diabetes (MD) might be misdiagnosed as type 1 diabetes. The prevalence of MD among children with apparent type 1 diabetes has not been established. Our aim was to estimate the prevalence of common forms of MD in childhood diabetes.
Methods
We investigated 2,756 children aged 0–14 years with newly diagnosed diabetes who had been recruited to the nationwide population-based Norwegian Childhood Diabetes Registry (NCDR), from July 2002 to March 2012. Completeness of ascertainment was 91%. Children diagnosed with diabetes who were under12 months of age were screened for mutations in KCNJ11, ABCC8 and INS. Children without GAD and protein tyrosine phosphatase-like protein antibodies were screened in two ways. Those who had a parent with diabetes were screened for mutations in HNF1A, HNF4A, INS and MT-TL1. Children with HbA1c <7.5% (<58 mmol/mol) and no insulin requirement were screened for mutations in GCK. Finally, we searched the Norwegian MODY Registry for children with genetically verified MD.
Results
We identified 15 children harbouring a mutation in HNF1A, nine with one in GCK, four with one in KCNJ11, one child with a mutation in INS and none with a mutation in MT-TL1. The minimum prevalence of MD in the NCDR was therefore 1.1%. By searching the Norwegian MODY Registry, we found 24 children with glucokinase-MODY, 15 of whom were not present in the NCDR. We estimated the minimum prevalence of MD among Norwegian children to be 3.1/100,000.
Conclusions/interpretation
This is the first prevalence study of the common forms of MD in a nationwide, population-based registry of childhood diabetes. We found that 1.1% of patients in the Norwegian Childhood Diabetes Registry had MD.
Similar content being viewed by others

Introduction
Monogenic diabetes (MD) is a heterogeneous group of rare diabetes forms in which a single mutation in one of more than 20 genes leads to decreased insulin secretion or severe insulin resistance [1, 2]. MD is commonly divided into permanent neonatal diabetes mellitus (PNDM), when the child is diagnosed with diabetes before 6 months of age, and MODY, when diabetes is diagnosed between 6 months and 25 years of age. MD also includes maternally inherited diabetes and deafness (MIDD) and some rare syndromes of insulin resistance.
Mutations in the genes KCNJ11, ABCC8 and INS are most common in PNDM [3–5]. In populations where consanguinity is often seen, mutations in EIF2AK3 and GCK are most frequent [6]. PNDM associated with syndromic features can be caused by a mutation in any of the genes PTF1A, RFX6, NEUROD1, FOXP3 and WFS1 [1].
MODY was initially a clinical diagnosis defined as autosomal dominant non-ketotic diabetes with an age of onset before 25 years. Recent gene discoveries have identified subtypes of MD that do not fit well with the classical criteria. However, the term MODY is still commonly used and 13 different genes have been associated with MODY [1, 7]. Approximately 70% of cases of MODY are due to mutations in GCK, HNF1A or HNF4A [8–10]. Glucokinase (GCK)-MODY is characterised by mild fasting hyperglycaemia. Pharmaceutical treatment is usually only necessary during pregnancy. There is no risk of diabetes-associated complications [11]. Hepatocyte nuclear factor 1α (HNF1A)-MODY and hepatocyte nuclear factor 4α (HNF4A)-MODY are phenotypically similar, presenting with progressive beta cell dysfunction and often sensitivity to sulfonylureas [12]. These two types of MODY are considered to be severe forms of diabetes, having an occurrence of late diabetic complications comparable to that of type 1 diabetes [13].
Some types of MODY are associated with extrapancreatic and/or systemic features, such as urogenital and pancreatic malformations in renal cysts and diabetes syndrome (also named HNF1B-MODY) [14, 15], and pancreatic exocrine dysfunction in carboxyl ester lipase (CEL)-MODY [16]. These features can be used to narrow the search for a correct diagnosis [17]. Mutations in PDX1, NEUROD1, KLF11, PAX4, INS, BKL, ABCC8 and KCNJ11 cause other rare forms of MODY [1].
MIDD is a syndrome caused by a mutation in the MT-TL1 gene in the mitochondria. The mean age at diagnosis of diabetes has been shown to be between 30 and 40 years, with a range of 11–68 years [18]. Several mutations in the MT-TL1 gene are known to cause diabetes, but the m.3243A<G mutation is by far the most common, being responsible for more than 80% of all cases [19].
The true prevalence of MD is not known, but estimates indicate that it may comprise 1–4% of all cases of diabetes [8, 10, 20–27]. MD might be misdiagnosed and hence treated as type 1 diabetes in children and possibly as type 2 diabetes in adults. Therefore, diagnosing MD can lead to a change in treatment, often with improved glycaemic control, not only for the individual, but also for their relatives [28, 29]. Moreover, a molecular diagnosis of MD is important for appropriate genetic counselling [2].
Our aim was to estimate the prevalence of MD among Norwegian children with diabetes. To this end, we screened genes causing common forms of MD in patients from the Norwegian Childhood Diabetes Registry (NCDR) and linked this registry with the Norwegian MODY Registry.
Methods
Study population
Since July 2002, all Norwegian children developing diabetes have been invited to participate in the NCDR. The completeness of ascertainment of this registry has been calculated to be 91% (T Skrivarhaug, L. C. M. Stene, H. Strøm, A. K. Drivvoll, P. R. Njølstad, G. Joner, unpublished data). The registry contains clinical data regarding onset of diabetes, family history and treatment, as well as serum and DNA samples. By 1 April 2012, the registry had included 2,756 children, all of whom had been younger than 15 years of age when diagnosed.
Screening methods
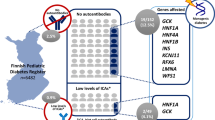
We selected three subsets of participants for genetic screening: (1) the PNDM group: all children with an onset of diabetes before 12 months of age (Fig. 1); (2) the MODY A group: all children who had at least one parent with diabetes, and who were GAD- and protein tyrosine phosphatase-like protein (IA2)-negative (Fig. 2); and (3) the MODY B group: all children who were GAD- and IA2-negative and had an HbA1c <7.5% (<58 mmol/mol) and no insulin requirement at hospital discharge (Fig. 3). The criteria for autoantibody negativity were an antibody index (AI) <0.08, or a concentration <1.0 U/ml of GAD and an AI <0.1, or a concentration <1.0 U/ml of IA2. We screened the PNDM group for mutations in KCNJ11, ABCC8 and INS, the MODY A group for mutations in HNF1A, HNF4A, INS and for the mutation m.3243A<G in the MT-TL1 gene, and the MODY B group for mutations in GCK (Figs 1–3).

Flowchart illustrating the screening for mutations in permanent neonatal diabetes genes. Among the 2,756 children in the Norwegian Childhood Diabetes Registry, 24 children were diagnosed with diabetes before 12 months of age. KCNJ11, ABCC8 and INS sequencing were performed for all these children, except for two (DNA unavailable). A KCNJ11 gene mutation was found in four children whereas no mutations were found in the remaining 18 children

Flowchart illustrating the screening for mutations in HNF1A, HNF4A, INS and MT-TL1. Among the 2,756 children in the Norwegian Childhood Diabetes Registry, antibody status (IA2 and GAD) was known in 2,525. Of the 323 children that were antibody-negative, 46 had a parent with diabetes and underwent HNF1A, HNF4A, INS and MT-TL1 sequencing. An HNF1A mutation was found in five individuals and an INS mutation in one child

Flowchart illustrating the screening for mutations in GCK. Among the 2,756 children in the Norwegian Childhood Diabetes Registry, antibody status (IA2 and GAD) was known in 2,525 children. Of the 323 children that were antibody-negative, 23 had HbA1c level at diagnosis <7.5% (58 mmol/mol). Of these, ten were discharged from hospital without insulin treatment and underwent GCK sequencing. A GCK mutation was found in three children
Linking the NCDR and the Norwegian MODY Registry
The Norwegian MODY Registry was established in 1997 and is a national, population-based registry. Physicians refer patients for genetic screening using the following criteria for the patient: (1) a first-degree relative with diabetes; (2) an onset of diabetes before age 25 years in at least one family member; (3) a low-dose insulin requirement; and (4) unusual type 1 diabetes (an insulin requirement below 0.5 U kg−1 day−1, no antibodies or an atypical history). A patient does not necessarily need to meet all the criteria for referral.
Depending on the clinical presentation, the patient is screened for mutations in one or more of the following genes: HNF4A, GCK, HNF1A, HNF1B and INS. Furthermore, all children with an onset of diabetes before 9 months of age are routinely referred from the NCDR to the Norwegian MODY Registry. They are tested for mutations in one or more of the following genes: KCNJ11, ABCC8 and INS. The registry also conducts predictive testing of non-diabetic individuals for mutations known to be present in their families. By 1 April 2012, the MODY registry had included 689 families. The mean age of patients when referred to the registry is 35 years (SD 19 years). We made a systematic search for patients appearing in both registries. Information regarding genetic diagnosis was retrieved from the Norwegian MODY Registry.
Genetic analyses
Genomic DNA was extracted from peripheral leucocytes using standard procedures. The coding exons and intron/exon boundaries of HNF1A, HNF4A, GCK, HNF1B, ABCC8, KCNJ11and INS (including the intronic c.188-31G>A mutation [30]) were amplified by PCR and sequenced using an Applied Biosystems 3730 capillary sequencer (Applied Biosystems, Foster City, CA). Primers and reaction conditions are available on request. The sample files were imported into the SeqScape Software (Applied Biosystems) and analysed for sequence variations by comparing with published reference sequences (NM_000545.5, NM_175914.3, NM_000162.3, NM_000458.2, NM_000352.3, NM_000525.3 and NM_000207.2, respectively). We also investigated for the presence of the mutation m.3243A>G in the mitochondrial tRNA leucine 1 gene (MT-TL1; NC_012920.1).
Laboratory analyses
GAD and IA2 were analysed at Haukeland University Hospital or Oslo University Hospital, Aker. Haukeland University Hospital used a radio ligand assay in which recombinant anti-GAD and anti-IA2 were labelled with 125I. Results are given in units per millilitre. At the Oslo University Hospital, a different radio ligand assay was used in which anti-GAD and anti-IA2 were labelled with 3H. Results are given as an AI. In both cases, the tracers were used in excess and bound by the respective antibodies.
Statistical analysis
A minimum prevalence of MD among the Norwegian child population aged 0–14 years was calculated by counting all children with MD who were younger than 15 years of age on 1 April 2012. By using numbers from Statistics Norway (www.ssb.no), we were able to estimate the total population of children aged 0–14 years on 1 April 2012 to be 880,000. The prevalence of permanent neonatal diabetes among live births was calculated in the same way. Inclusion was, however, limited to 1 April 2011 since all children had to be observed for at least 1 year to exclude transient neonatal diabetes. In the period from 1 July 2002 to 1 April 2011, 513,687 children were born in Norway.
Ethics
The present study was conducted in accordance with the Declaration of Helsinki and approved by the Regional Committee for Research Ethics. Written informed consent was obtained from all patients/guardians.
Results
PNDM in the NCDR
We identified 24 children in the PNDM group. All had permanent diabetes at follow-up at least 1 year after diagnosis (mean age 4 years, range 1–8 years). Of these, four children had been diagnosed before 6 months of age. Thus, 0.15% (4/2,756) of the children in the NCDR had neonatal diabetes. DNA was available for 22/24 children (Fig. 1). Four children had a pathogenic mutation in the KCNJ11 gene. They all had the same de novo mutation, c.175G>A/p.Val59Met, and had been diagnosed with diabetes at 1, 2, 4 and 4 months of age, respectively (Table 1). We observed no children with mutations in ABCC8 or INS.
MODY and MIDD in the NCDR
We excluded 231 of the 2,756 children in the NCDR due to a lack of information regarding their autoantibody status. Among the 2,525 children who had autoantibodies analysed, 323 were negative for GAD and IA2. Of these, 46 had a parent with diabetes and thus met our screening criteria (Fig. 2). Sequencing of HNF1A, HNF4A and INS in these 46 children revealed five individuals carrying mutations in HNF1A and one child with a mutation in INS (Table 1). None had the m.3243A<G mutation in MT-TL1.
Among the 323 children who were negative for GAD and IA2, 23 children had an HbA1c <7.5% (<58 mmol/mol) at diagnosis. Of these, ten had no insulin requirement at discharge from hospital and thus met our screening criteria (Fig. 3). We screened for GCK in these ten children and found three children harbouring a mutation (Table 1).
Because GCK-MODY has a mild phenotype, we looked into why diabetes had been recognised in these three individuals. One child presented with symptoms of fatigue and a set of blood samples showed elevated glucose levels. The family history revealed several members with apparent type 2 diabetes. The child’s fatigue disappeared without treatment. The second child had a family where several members had diet-regulated diabetes and a home measurement of blood glucose had shown elevated levels. The last child had a mother with a known GCK-MODY.
Linking the NCDR and the Norwegian MODY Registry
We identified 88 children who appeared in both registries. One child harbouring a mutation in ABCC8 was excluded since he had been pancreatectomised due to congenital hyperinsulinism of infancy [31]. Of the 87 remaining children, 30 had a genetic diagnosis in the Norwegian MODY Registry. Of these, 15 harboured a mutation in HNF1A, nine a mutation in GCK, four a mutation in KCNJ11, one a mutation in INS and one a mutation in CEL (Table 1). Hence, the minimum prevalence of MD in the NCDR was 1.1% (30/2,756).
GCK-MODY in the Norwegian MODY Registry
Due to the mild phenotype, children with GCK-MODY are rarely referred to hospital clinics. Since recruitment to the NCDR is done by hospital paediatricians, this might lead to an underreporting of GCK-MODY to the NCDR. To assess this possibility, we searched the Norwegian MODY Registry for children (0–14 years at referral) with a genetic diagnosis of GCK-MODY. We identified 24 patients, 15 of whom had not been reported to the NCDR.
Prevalence of MD in the Norwegian child population
By 1 April 2012, we had found 27 children below the age of 15 years with a diagnosis of MD. Of these, 19 children had a mutation in the GCK gene, four had a mutation in the HNF1A gene and four a mutation in the KCNJ11 gene. The minimum prevalence of MD in the total Norwegian child population below 15 years of age was therefore 3.1/100,000 (27/880,000).
Prevalence of genetic MD diagnosis in the Norwegian child population
By 1 April 2012, we had found 49 children below the age of 15 years with a genetic MD diagnosis in the Norwegian MODY Registry. Of these, 19 had a mutation in the GCK gene, 12 had a mutation in the HNF1A gene, six had a mutation in the HNF1B gene, eight had a mutation in the CEL gene and four had a mutation in the KCNJ11 gene. At the time of referral to the MODY Registry, 22 of the 49 children had not developed diabetes. The minimum prevalence of children with a genetic MD diagnosis in the total Norwegian child population below 15 years of age was therefore 5.6/100,000 (49/880,000).
Discussion
To our knowledge, this is the first study to estimate the prevalence of MD in a nationwide population of children with diabetes. In our study, the minimum prevalence of MD in 2,756 children with newly diagnosed diabetes was 1.1%.
We found the prevalence of PNDM to be 0.15% among children with diabetes, or 1/130,000 live births. This is comparable to an Italian study that estimated the prevalence of PNDM to be 1/152,000 (16 affected children) [20]. A Slovakian study found the prevalence of PNDM to be 1/215,000 live births (eight affected children) [21]. Studies from USA, the UK, the Netherlands and Poland have reported a prevalence of about 1/250,000 live births [22, 23].
The four children with PNDM in our study all had mutations in the KCNJ11 gene (Table 1), compared with seven of 16 and four of eight in the Italian and Slovakian studies, respectively. In the US study, two children had mutations in KCNJ11, whereas mutations in INS were found in three children. Although the numbers are small, this indicates some difference in the prevalence and aetiology of PNDM across populations. The difference is even more striking when looking at populations where consanguinity is relatively common [6].
When retrieving clinical and family data from the 24 children in the PNDM group, we did not reveal any information pointing towards children with parents who were closely related or children with syndromic PNDM. By searching the Norwegian MODY Registry, we found two children with homozygous mutations in the GCK gene. These were not included in the NCDR since they had developed diabetes before the NCDR had started including cases.
Mutations in the insulin receptor gene might lead to the Donohue or Rabson–Mendenhall syndromes, where onset of diabetes is in the neonatal period (OMIM *147670). These syndromes are associated with severe insulin resistance and, especially Donohue syndrome, show high mortality. To assess the possibility that some of the children in the NCDR had severe insulin resistance, we looked at the levels of C-peptide at the time of diagnosis and the insulin requirement at discharge from hospital. Among the 1,117 children whose C-peptide level was analysed, the highest reported value was 2.3 nmol/l (normal fasting range: 0.22–1.4 nmol/l). Although C-peptide results should be interpreted with caution, cases with severe insulin resistance usually have highly elevated C-peptide levels, indicating that there are no cases in our material. Looking at the children’s insulin treatment when discharged from hospital, the highest insulin dose was 3 U kg−1 day−1. Although higher than the usual 0.8 U kg−1 day−1, this insulin requirement is lower than that in severe insulin resistance, where doses up to 3–5 U kg−1 day−1 are necessary. Furthermore, these cases all had low C-peptide levels, indicating some sort of beta cell dysfunction.
We found a minimum prevalence of MODY of 0.94% among children with diabetes (26 children). This is somewhat higher than previously reported figures for prevalence, especially when taking into account that our population is younger than that of comparable studies, and that many children with a pathogenic mutation in an MODY gene develop diabetes after the age of 15 years. The prevalence of clinical and genetic MODY reported in a German/Austrian study of 40,757 children and adolescents diagnosed with diabetes was 0.83% and 0.65%, respectively [10]. The Norwegian HUNT2 study tested 1,850 Norwegian adults with diabetes for three mutations in HNF1A, establishing a minimum prevalence of 0.4%. Clinical criteria for MODY were fulfilled in 2.2% [25]. A cross-sectional study in Oxfordshire, UK, found a minimum prevalence of HNF1A-MODY of 0.18% among young adults with diabetes [26]. A UK study of referral patterns related to genetic testing for MODY in the UK indicated a minimum population prevalence of MD of 108 per million [8].
The estimated prevalence of MD in the Norwegian child population under 15 years of age was 3.1/100,000. This is lower than in a Polish study that reported a prevalence of 4.6/100,000 [27]. The latter study was, however, based on children and youth less than 18 years of age. Furthermore, a high proportion of GCK-MODY (83%) was reported, indicating a different distribution of MD genes.
We found no cases of MIDD in the NCDR. Several studies have shown the prevalence of MIDD among adults with diabetes to be 1% [18]. In MIDD, diabetes usually develops after the age of 30 years, which may explain why we did not find any cases in our material. HNF1A-MODY was the most prevalent MODY subtype in our study, accounting for 15 of 26 cases (58%). Nine of 26 children (35%) had GCK-MODY. Mutations in INS were less common. Knowing the mild phenotype of GCK-MODY, there might be an underreporting to the NCDR. Given that the hyperglycaemia seen in GCK-MODY starts at a young age (from birth), the finding of an additional 15 children with GCK-MODY in the Norwegian MODY Registry who were not present in the NCDR supports this assumption. Including these cases, the proportion of GCK-MODY would then be 59% (24/41), while that of HNF1A-MODY would be 37% (15/41).
Our study has limitations. We used Sanger sequencing when screening for MODY and PNDM. Testing for possible large pathogenic deletions and genomic rearrangements might increase the detection rate of MD as these are not detected by direct DNA sequencing. Among families with MODY who were negative for mutations in HNF1A and HNF4A, 7.1% had a deletion of part of or the complete HNF4A gene, whereas 2.4% showed this for the HNF1A gene [32]. When screening for MODY, we used a family history of diabetes as a selection criterion. This was reported as either positive (a parent with diabetes) or negative. In the negative group, there were children with parents without diabetes, but also children for whom their parents’ diabetes status was unknown. Thus, among the 277 children reported to have no diabetes in their families (Fig. 2), there could be some who actually have a positive family history and therefore should have been screened. Furthermore, 231 children did not have GAD or IA2 analysed and were omitted from genetic analysis (Fig. 2). Elevated levels of IA2 and GAD autoantibodies indicate type 1 diabetes. By linking the two registries, we found three children with MODY who had positive autoantibodies (Table 1). This might indicate that some children with positive autoantibodies, albeit in low titres, might have MD. Since MODY in children and adolescents can clinically resemble type 1 or 2 diabetes, cases might be hard to find. Different knowledge among clinicians and services for genetic testing and counselling might explain some of the variations seen. This was shown in the UK, where the referral rates correlated well with different funding of genetic analyses [8].
What is the maximum prevalence of MD in Norwegian children with diabetes? Making this estimate is not straightforward, and different approaches might give significantly different answers. Our approach starts with an assumption that all children negative for GAD and IA2 are ‘possible MD’ cases. This is, however, not entirely true since a proportion of the healthy population are GAD or IA2 positive [33], and this could also be the case for some patients with MD. On the other hand, some cases of type 1 diabetes are GAD and IA2 negative at the onset of diabetes. Furthermore, children with type 2 diabetes are also GAD and IA2 negative. In assessing this problem, we used a BMI greater than +2 SD adjusted for age and sex as a criterion to find those children most likely to have type 2 diabetes. We found that 323 of 2,525 children were negative for GAD or IA2. Among these 323, we found 16 children with a BMI above +2 SD, and thus more likely to have type 2 diabetes. With this adjustment, the maximum prevalence of MD among Norwegian children with diabetes is 12% (307/2,525).
In this study, we report a minimum prevalence of 1.1% among children with diabetes and 3.1/100,000 in the child population. The prevalence of a genetic diagnosis of MD in the Norwegian child population is 5.6/100,000. The true prevalence is higher, considering the underreporting of GCK-MODY, but we believe that it is a good estimate of MD with a more severe phenotype, since the screening was performed using a national- and population-based registry with a high degree of ascertainment. The exact prevalence of MD among children will, however, remain unknown until all relevant MD genes are screened in population-based studies, for instances by whole-genome sequencing [34].
Abbreviations
- AI:
-
Antibody index
- CEL :
-
Carboxyl ester lipase
- GCK :
-
Glucokinase
- HNF1A:
-
Hepatocyte nuclear factor 1α
- IA2:
-
Protein tyrosine phosphatase-like protein
- MD:
-
Monogenic diabetes
- MIDD:
-
Maternally inherited diabetes and deafness
- NCDR:
-
Norwegian Childhood Diabetes Registry
- PNDM:
-
Permanent neonatal diabetes mellitus
References
Molven A, Njolstad PR (2011) Role of molecular genetics in transforming diagnosis of diabetes mellitus. Expert Rev Mol Diagn 11:313–320
Vaxillaire M, Bonnefond A, Froguel P (2012) The lessons of early-onset monogenic diabetes for the understanding of diabetes pathogenesis. Best Pract Res Clin Endocrinol Metab 26:171–187
Gloyn AL, Pearson ER, Antcliff JF et al (2004) Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med 350:1838–1849
Babenko AP, Polak M, Cave H et al (2006) Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N Engl J Med 355:456–466
Stoy J, Steiner DF, Park SY, Ye H, Philipson LH, Bell GI (2010) Clinical and molecular genetics of neonatal diabetes due to mutations in the insulin gene. Rev Endocr Metab Disord 11:205–215
Habeb AM, Flanagan SE, Deeb A et al (2012) Permanent neonatal diabetes: different aetiology in Arabs compared to Europeans. Arch Dis Child 97:721–723
Bonnefond A, Philippe J, Durand E et al (2012) Whole-exome sequencing and high throughput genotyping identified KCNJ11 as the thirteenth MODY gene. PLoS One 7:e37423
Shields BM, Hicks S, Shepherd MH, Colclough K, Hattersley AT, Ellard S (2010) Maturity-onset diabetes of the young (MODY): how many cases are we missing? Diabetologia 53:2504–2508
Estalella I, Rica I, Perez de Nanclares G et al (2007) Mutations in GCK and HNF-1alpha explain the majority of cases with clinical diagnosis of MODY in Spain. Clin Endocrinol 67:538–546
Schober E, Rami B, Grabert M et al (2009) Phenotypical aspects of maturity-onset diabetes of the young (MODY diabetes) in comparison with type 2 diabetes mellitus (T2DM) in children and adolescents: experience from a large multicentre database. Diabet Med J Br Diabet Assoc 26:466–473
Page RC, Hattersley AT, Levy JC et al (1995) Clinical characteristics of subjects with a missense mutation in glucokinase. Diabet Med J Br Diabet Assoc 12:209–217
Shepherd M, Shields B, Ellard S, Rubio-Cabezas O, Hattersley AT (2009) A genetic diagnosis of HNF1A diabetes alters treatment and improves glycaemic control in the majority of insulin-treated patients. Diabet Med J Br Diabet Assoc 26:437–441
Isomaa B, Henricsson M, Lehto M et al (1998) Chronic diabetic complications in patients with MODY3 diabetes. Diabetologia 41:467–473
Bellanne-Chantelot C, Chauveau D, Gautier JF et al (2004) Clinical spectrum associated with hepatocyte nuclear factor-1beta mutations. Ann Intern Med 140:510–517
Oram RA, Edghill EL, Blackman J et al (2010) Mutations in the hepatocyte nuclear factor-1β (HNF1B) gene are common with combined uterine and renal malformations but are not found with isolated uterine malformations. Am J Obstet Gynecol 203(364):e1–e5
Raeder H, Johansson S, Holm PI et al (2006) Mutations in the CEL VNTR cause a syndrome of diabetes and pancreatic exocrine dysfunction. Nat Genet 38:54–62
Haldorsen IS, Raeder H, Vesterhus M, Molven A, Njolstad PR (2012) The role of pancreatic imaging in monogenic diabetes mellitus. Nat Rev Endocrinol 8:148–159
Murphy R, Turnbull DM, Walker M, Hattersley AT (2008) Clinical features, diagnosis and management of maternally inherited diabetes and deafness (MIDD) associated with the 3243A>G mitochondrial point mutation. Diabet Med J Br Diabet Assoc 25:383–399
Tsukuda K, Suzuki Y, Kameoka K et al (1997) Screening of patients with maternally transmitted diabetes for mitochondrial gene mutations in the tRNA[Leu(UUR)] region. Diabet Med J Br Diabet Assoc 14:1032–1037
Iafusco D, Massa O, Pasquino B et al (2012) Minimal incidence of neonatal/infancy onset diabetes in Italy is 1:90,000 live births. Acta Diabetol 49:405–408
Stanik J, Gasperikova D, Paskova M et al (2007) Prevalence of permanent neonatal diabetes in Slovakia and successful replacement of insulin with sulfonylurea therapy in KCNJ11 and ABCC8 mutation carriers. J Clin Endocrinol Metab 92:1276–1282
Slingerland AS, Shields BM, Flanagan SE et al (2009) Referral rates for diagnostic testing support an incidence of permanent neonatal diabetes in three European countries of at least 1 in 260,000 live births. Diabetologia 52:1683–1685
Shankar RK, Pihoker C, Dolan LM et al (2012) Permanent neonatal diabetes mellitus: prevalence and genetic diagnosis in the SEARCH for Diabetes in Youth Study. Pediatr Diabetes. doi:10.1111/pedi.12003
Wiedemann B, Schober E, Waldhoer T et al (2010) Incidence of neonatal diabetes in Austria—calculation based on the Austrian Diabetes Register. Pediatr Diabetes 11:18–23
Eide SA, Raeder H, Johansson S et al (2008) Prevalence of HNF1A (MODY3) mutations in a Norwegian population (the HUNT2 Study). Diabet Med J Br Diabet Assoc 25:775–781
Kropff J, Selwood MP, McCarthy MI, Farmer AJ, Owen KR (2011) Prevalence of monogenic diabetes in young adults: a community-based, cross-sectional study in Oxfordshire, UK. Diabetologia 54:1261–1263
Fendler W, Borowiec M, Baranowska-Jazwiecka A et al (2012) Prevalence of monogenic diabetes amongst Polish children after a nationwide genetic screening campaign. Diabetologia 55:2631–2635
Sagen JV, Raeder H, Hathout E et al (2004) Permanent neonatal diabetes due to mutations in KCNJ11 encoding Kir6.2: patient characteristics and initial response to sulfonylurea therapy. Diabetes 53:2713–2718
Pearson ER, Flechtner I, Njolstad PR et al (2006) Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N Engl J Med 355:467–477
Garin I, Perez de Nanclares G, Gastaldo E, Harries LW, Rubio-Cabezas O, Castano L (2012) Permanent neonatal diabetes caused by creation of an ectopic splice site within the INS gene. PLoS One 7:e29205
Sandal T, Laborie LB, Brusgaard K et al (2009) The spectrum of ABCC8 mutations in Norwegian patients with congenital hyperinsulinism of infancy. Clin Genet 75:440–448
Carette C, Dubois-Laforgue D, Saint-Martin C et al (2010) Familial young-onset forms of diabetes related to HNF4A and rare HNF1A molecular aetiologies. Diabet Med J Br Diabet Assoc 27:1454–1458
Schlosser M, Strebelow M, Wassmuth R et al (2002) The Karlsburg type 1 diabetes risk study of a normal schoolchild population: association of beta-cell autoantibodies and human leukocyte antigen-DQB1 alleles in antibody-positive individuals. J Clin Endocrinol Metab 87:2254–2261
Johansson S, Irgens H, Chudasama KK et al (2012) Exome sequencing and genetic testing for MODY. PLoS One 7:e38050
Acknowledgements
We thank all the personnel at the Norwegian MODY Registry and the NCDR for their support and interest in the study.
Funding
Funding was received from Western Norway Regional Authority (H. Irgens, P. R. Njølstad), University of Bergen (A. Molven, P. R. Njølstad), Haukeland University Hospital (H. Irgens, P. R. Njølstad), Innovest (P. R. Njølstad), the Norwegian Research Council (P. R. Njølstad), the European Research Council (P. R. Njølstad), and the KG Jebsen Foundation (P. R. Njølstad.).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript. The funders had no role in the study design, data collection and analysis, the decision to publish, or preparation of the manuscript.
Contribution statement
AM and PRN developed the study concept and supervised the study. HI researched and interpreted the data, analysed the data and drafted the first manuscript. GJ, TS, DEU and OS helped with the study design, and GJ provided statistical advice. JM, BBJ and MR carried out the genetic analyses and helped interpret the data. TS helped with data collection. All authors contributed to the interpretation of the results and to the discussion, reviewed the paper and approved the final version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Irgens, H.U., Molnes, J., Johansson, B.B. et al. Prevalence of monogenic diabetes in the population-based Norwegian Childhood Diabetes Registry. Diabetologia 56, 1512–1519 (2013). https://doi.org/10.1007/s00125-013-2916-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-013-2916-y