
Abstract
Aims/hypothesis
Dominantly acting loss-of-function mutations in the ABCC8/KCNJ11 genes can cause mild medically responsive hyperinsulinaemic hypoglycaemia (HH). As controversy exists over whether these mutations predispose to diabetes in adulthood we investigated the prevalence of diabetes in families with dominantly inherited ATP-sensitive potassium (KATP) channel mutations causing HH in the proband.
Methods
We studied the phenotype of 30 mutation carriers (14 children and 16 adults) from nine families with dominant ABCC8/KCNJ11 mutations. Functional consequences of six novel missense mutations were examined by reconstituting the KATP channel in human embryonic kidney 293 (HEK293) cells and evaluating the effect of drugs and metabolic poisoning on the channels using the 86Rb flux assay.
Results
The mutant channels all showed a lack of 86Rb efflux on exposure to the channel agonist diazoxide or metabolic inhibition. In the families, dominant ABCC8/KCNJ11 mutations were associated with increased birthweight (median + 1.56 SD score [SDS]). Fourteen children had HH and five adults were reported with HH or hypoglycaemic episodes (63%). Progression from hypoglycaemia to diabetes mellitus occurred in two individuals. Eight adults had a history of gestational diabetes in multiple pregnancies or were diabetic (diagnosed at a median age of 31 years). Within these families, none of the 19 adults who were not carriers of the ABCC8/KCNJ11 mutation was known to be diabetic.
Conclusions/interpretation
The phenotype associated with dominant ABCC8/KCNJ11 mutations ranges from asymptomatic macrosomia to persistent HH in childhood. In adults, it may also be an important cause of dominantly inherited early-onset diabetes mellitus.
Similar content being viewed by others

Introduction
Hyperinsulinaemic hypoglycaemia (HH) occurs because of inappropriate secretion of insulin by the pancreatic beta cells. The congenital forms of HH (CHI) occur because of defects in key genes involved in insulin secretion [1]. The commonest and most severe forms of CHI are due to mutations in the ABCC8 and KCNJ11 genes [2, 3], encoding the sulfonylurea receptor 1 (SUR1) and inward rectifier K(+) channel Kir6.2 (Kir6.2) subunits of the ATP-sensitive potassium (KATP) channel, respectively. The KATP channels play a pivotal role in glucose-stimulated insulin secretion and couple glucose metabolism to membrane electrical activity and insulin release in the pancreatic beta cells [4].
Recessively inherited inactivating ABCC8/KCNJ11 mutations usually cause medically unresponsive disease. The molecular basis of the CHI observed in these patients involves defects in the biogenesis and turnover of KATP channels, in the trafficking of channels to the plasma membrane and alterations in the open-state frequency through changes in nucleotide sensitivity [5–8].
Dominant ABCC8/KCNJ11 mutations are less frequently reported and generally cause mild medically responsive HH [9–12] or in rare cases, severe unresponsive HH [13]. Two reports suggest that medically responsive HH due to a dominantly inherited ABCC8 mutation may progress to diabetes mellitus (DM) in later life [9, 12, 14]. However, this was not supported in a recent case series where only 4 out of 29 adult mutation carriers developed DM [11]. Hence, it is not clear whether dominantly acting mutations in the genes that cause diazoxide-responsive HH in childhood are associated with later development of DM in adulthood.
In this study, we present the phenotype of eight different heterozygous ABCC8/KCNJ11 mutations in nine families and report the prevalence of DM in the adult mutation carriers. We show that dominant mutations can cause a variable phenotype ranging from asymptomatic macrosomia to transient/persistent HH as well as progression to DM in later life.
Methods
Patients
Nine families with dominant KATP channel mutations were studied. The probands are a subgroup of children referred to the tertiary Hyperinsulinism Service at Great Ormond Street Hospital NHS Trust, London, UK. They include those children with HH who were diazoxide responsive and were identified to be heterozygous for a KATP channel mutation. The diagnosis of HH was based on diagnostic criteria described previously (i.e. inappropriately elevated insulin concentrations at the time of hypoglycaemia with corresponding low concentrations of plasma β-hydroxybutyrate and fatty acids). Diazoxide responsiveness was defined as the ability to maintain normoglycaemia without the support of intravenous glucose. Clinical information (birthweight, age at presentation, treatment details of HH) was collected from the case notes and the referring clinicians. Family history was specifically explored with regards to symptoms of hypoglycaemia and presence/absence of DM and phenotypic details of individuals affected by hypoglycaemia/DM (birthweight, age of presentation, treatment details) was collected. The Office for National Statistics (ONS) classification was used to categorise ethnicity [15]. The study was approved by the regional ethical committee and written consent was obtained from the families.
Genetic analysis
Genomic DNA was extracted from peripheral leucocytes using standard procedures. The single exon of the KCNJ11 gene and the 39 exons of ABCC8 were amplified by PCR using previously reported primers [16]. Single-strand sequencing was carried out using standard methods and an ABI3730 capillary sequencer (Applied Biosystems, Warrington, UK) and sequences were compared with the published sequences (NM_000525 and NM_000352.2 [which incorporates the alternatively spliced amino acid in exon 17 {L78224}]) using Mutation Surveyor 3.24 software (SoftGenetics, State College, PA, USA). Following identification of a mono-allelic KATP channel mutation in the proband, parents were tested to establish the mode of inheritance. Microsatellite analysis (PowerPlex 16 System, Promega, Southampton, UK) was undertaken to confirm family relationships when a de novo mutation was identified. Genetic analysis was then offered to the siblings of the proband (parental consent obtained) and to the siblings and parents of the parent identified as heterozygous for the mutation. Co-segregation studies were then undertaken with the available phenotypic data and results of the genetic analysis.
Functional analysis of mutant channels
Functional analysis of the novel ABCC8 mutations (A1508P, A1537V, L1431F, L1390R and Q1459E) and the KCNJ11 mutation (I284del) was carried out. Mutations were introduced in hamster Sur1 (also known as Abcc8) cDNA and mouse Kir6.2 (also known as Kcnj11) cDNA using the Quik-change site-directed mutagenesis kit (Agilent Technologies, Stockport, UK). Mutant KATP channel was reconstituted in human embryonic kidney 293 (HEK293) cells by transfection using FuGene HD (Roche, Burgess Hill, UK) according to the manufacturer’s instructions. Enhanced green fluorescent protein, 50 ng, was also co-transfected to determine transfection efficiency.
Following successful transfection, an 86Rb flux assay was used to determine functionality of the reconstituted channels as described previously by Muzyamba et al. [17]. The 86Rb efflux assay is based on the principle that when cells are loaded with rubidium, its distribution between intracellular and extracellular spaces is an indicator of channel activity as 86Rb is similar in size and charge to K+ and is handled by the channels in a manner similar to K+. Briefly, 24 h after transfection cells were incubated overnight with minimum essential medium (MEM) containing 86Rb (0.037 MBq/ml) at optimum conditions (5% CO2, 37°C). The assay medium was then aspirated and the cells were washed twice with 2 ml HEPES buffered NaCl (HBS) (10 mmol/l HEPES, 10 mmol/l glucose, 130 mmol/l NaCl, 7 mmol/l KCl, 2 mmol/l CaCl2 and 1 mmol/l MgCl2, pH 7.4) [17]. The cells were then incubated with 2 ml of HBS medium with or without channel stimulants and/or inhibitors. The following conditions were used: control cells (no drugs); cells treated with 100 μmol/l diazoxide; cells treated with 100 μmol/l diazoxide plus 10 μmol/l glibenclamide; cells treated with 2.5 mmol/l NaCN (sodium cyanide) and 20 mmol/l 2-deoxy-d-glucose; and cells treated with 2.5 mmol/l NaCN, 20 mmol/l 2-deoxy-d-glucose and 10 μmol/l glibenclamide. The concentrations of the drugs stated have been shown to be effective on this cell line [17]. The supernatant fraction was aspirated after 5 min into scintillation vials for counting and the cells were then lysed with 2% Triton/HBS solution. Cherenkov radiation in the supernatant fraction and cell lysates was measured and the efflux was calculated as a percentage relative to the total amount of radioactivity incorporated. The data are presented as mean ± SEM. Significant differences were tested by employing the one-way ANOVA and Bonferroni post-test. Significant differences are reported as: *p ≤ 0.05, **p ≤ 0.01 and ***p ≤ 0.001.
The level of mutant protein produced in the transfected cells was detected using western blotting. The antibody anti-SUR1 NBD2 was raised in sheep to the peptide epitope ETLLSQKDSVFASFVRADK (University of Dundee, Dundee, UK) and subsequently used as described previously by Muzyamba et al. [17]. An anti-sheep IgG secondary antibody (Sigma, Poole, UK) linked to horseradish peroxidase and the ECL detection kit (GE Healthcare, Amersham, UK) were used to detect protein production.
OGTT and BMI
The adult mutation carriers were divided into those with a history of DM (gestational or other, n = 8) and those without (n = 8). A 2 h OGTT was conducted in the relatives of adult mutation carriers who had no history of DM to ensure that undiagnosed impaired glucose tolerance/DM was detected. Patients with ongoing hypoglycaemia (II-2, family A and III-2, family F [Fig. 1]) were excluded. Six adults met the criteria but one participant (II-1, family E [Fig. 1]) was not contactable. Blood glucose concentrations were measured at 0 and 120 min following an oral load of glucose (75 g) in the remaining five adult mutation carriers. The BMI of the adult mutation carriers with normal glucose tolerance (n = 5) and with DM (n = 8) was calculated and the means were compared using the Student’s t test.

Partial pedigrees showing inheritance of the dominant mutations in the ten families. Circles represent females and squares indicate males. An arrow indicates the proband. Vertical hatching denotes diabetic individuals and diagonal hatching represents individuals with GDM. Black symbols denote children/adults with hypoglycaemia. Individuals who progressed from hypoglycaemia to diabetes are indicated by symbols that are half-filled and half-hatched vertically. The genotype is given below each symbol. M/N denotes a heterozygous ABCC8/KCNJ11 mutation and N/N a normal genotype. The KCNJ11 mutation is indicated by *; the remaining are ABCC8 gene mutations
Results
Genetic analysis
Seven different heterozygous ABCC8 mutations were identified in eight probands. Five of the mutations were novel (L1390R, L1431F, Q1459E, A1508P and A1537V) and two have been reported previously in patients with dominantly inherited HH (G1479R and R1539Q) [11]. Each mutation was identified in a single family with the exception of G1479R, which was identified in two unrelated families. All mutations affected residues that are highly conserved across species and occurred within nucleotide-binding domain 2 of SUR1, a previously reported hotspot for dominantly acting mutations. The remaining proband was heterozygous for an in-frame deletion (I284del) in the KCNJ11 gene.
Of the 35 adult family members (including 18 parents) and eight siblings screened for the identified mutation, 16 adults and five siblings tested positive. This included eight parents, of whom seven were affected with hypoglycaemia or DM. In the remaining case (family C) although the mutation (G1479R) had been inherited from the unaffected father it was shown to co-segregate with HH in the family having been identified in the proband’s two affected siblings and paternal cousin. In two families there was no history of hypoglycaemia or DM (Fig. 1, family I and J). Confirmation of family relationships by microsatellite analysis demonstrated that the L1390R and G1479A mutations had arisen de novo in the proband.
Functional analysis of mutant channels
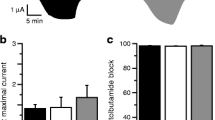
The R1539Q and G1479R ABCC8 mutations have been characterised previously by Pinney et al. [11] and hence these mutations were excluded from functional studies. The 86Rb flux assay showed that the channels with the mutant proteins (SUR1 [A1508P, L1431F, L1390R, Q1459E or A1537V] or Kir6.2 [I284del]) were non-functional as evidenced by the lack of 86Rb efflux on exposure to the channel agonist diazoxide or metabolic inhibition (Fig. 2a–g). Production of the mutant SUR1 proteins L1431F, L1390R, Q1459E and A1537V did not differ from the wild-type SUR1 protein on western blot analysis (Fig. 2h).

a–g Effects of SUR1/Kir6.2 mutants on KATP channel function studied with 86Rb efflux. The 86Rb efflux was assayed 48 h after transfection in control cells (horizontal hatching, no drugs), cells treated with 100 μmol/l diazoxide (diagonal hatching), cells treated with 100 μmol/l diazoxide plus 10 μmol/l glibenclamide (vertical hatching), cells treated with 2.5 mmol/l NaCN and 20 mmol 2-deoxy-d-glucose (black) and cells treated with 2.5 mmol/l NaCN, 20 mmol/l 2-deoxy-d-glucose and 10 μmol/l glibenclamide (white). (a) Wild type; (b) L143F (ABCC8); (c) A1508P (ABCC8); (d) I284del (KCNJ11); (e) L1390R (ABCC8); (f) Q1459E (ABCC8); (g) A1537V (ABCC8). Data represent means ± SEM for three different experiments performed in triplicate (n = 9); ***p < 0.001. h Western blot analysis of SUR1 shows that the production of the mutant SUR1 proteins L1431F, L1390R, Q1459E and A1537V did not differ from the WT SUR1 protein (177 kDa). WT, wild type
Clinical characteristics of children with HH
The clinical characteristics of the 14 children with HH (nine probands and five affected siblings) are summarised in Table 1. The median age at presentation was 1 day and median birthweight was +2.21 SD score (SDS). Hypoglycaemia resolved within 1 week in one case but the remaining children (aged 1–11 years) received ongoing diazoxide treatment.
Phenotype of the adult mutation carriers
The median birthweight of the adult mutation carriers (n = 16; data available for ten individuals) was high at +1.39 SDS (ranging from −2.11SDS to +2.45SDS, Table 2). Interestingly, five of the six adult mutation carriers who were macrosomic at birth developed hypoglycaemia. Two of these had persisting hypoglycaemia at 28 and 39 years of age (Fig. 1: family A, II-2 and family F, III-2), whilst one had a history of transient neonatal hypoglycaemia (Fig. 1: family E, II-1) (Table 2). Two other adults had a past history of hypoglycaemia that progressed to DM (family B II-2 and family H III-2).
Of the remaining 11 adult mutation carriers, there was a history of diabetes (gestational or other) in six further individuals (family A, II-3; family D, II-2; family G, III-5; family F, II-4 and III-4; and family H, II-2). The remaining five adults (family A I-2, family C I-2, II-2, II-3 and family F III-3) had no history of hypoglycaemia/DM. In total, 5/16 adult mutation carriers had or had had hypoglycaemia and eight either had gestational diabetes (GDM) in multiple pregnancies or were currently diabetic. The age at diagnosis of DM ranged from 20 to 50 years with a median of 31 years (see Table 2). None of the 19 adult family members (aged 20–68 years, median 40 years) who did not carry the ABCC8/KCNJ11 mutation were known to be affected with diabetes (Fig. 1).
According to the WHO criteria none of the five asymptomatic adult mutation carriers investigated had impaired fasting glucose/impaired glucose tolerance or DM. There was no significant difference in the mean BMI of the adult mutation carriers who developed DM from those who did not (p = 0.7).
Discussion
This case series reports the phenotype of eight (six novel) dominant ABCC8/KCNJ11 mutations. We identified 30 individuals (14 children and 16 adults) with heterozygous KATP channel mutations from ten families. Nineteen of the 30 (63%) had a past/current history of hypoglycaemia and eight adults (50%) had a history of gestational diabetes in multiple pregnancies or were currently diabetic.
The hyperinsulinaemic phenotype of these mutations was mild and diazoxide responsive, as reported in the literature previously [9, 11, 12]. Of the 14 children, 13 required treatment with diazoxide, whilst the remaining had transient hypoglycaemia that remitted in the neonatal period. These included a pair of siblings (family F [Fig. 1]) heterozygous for the R1539Q mutation in the ABCC8 gene who had hypoglycaemia for different durations. The older sibling required brief treatment with intravenous glucose after birth while the younger sibling continued to require diazoxide therapy at 3 years of age, demonstrating the variability in the phenotype of HH within the same family.
Of the 16 adult mutation carriers, two were recognised to have had hypoglycaemic episodes after the diagnosis of HH in their children. Three others had a previous history of hypoglycaemia; of these individuals, one had had transient neonatal hypoglycaemia treated with intravenous glucose, while the other two had had more persistent hypoglycaemia that remitted in young adulthood. The remaining adult mutation carriers (11/16) had no current/past history of hypoglycaemia. This is consistent with the study by Pinney et al. in which 14/29 adults were asymptomatic [11]. The incomplete penetrance of hypoglycaemia is not mutation specific. Nevertheless, in utero hyperinsulinism was evident from the higher birthweights of the mutation carriers. Hence, these dominant K ATP mutations result in a varied phenotype, ranging from asymptomatic macrosomia to persistent HH requiring diazoxide treatment. In contrast to the paper by Huopio et al. [9], in which all the patients inherited their mutation from their mothers, our probands inherited the mutations from either the maternal or paternal allele, refuting the possibility of a parent-of-origin effect or an imprinting defect.
It has been debated whether dominant KATP channel mutations causing HH predispose to the development of DM in adulthood [9, 11, 14, 18]. Huopio et al. first reported a dominant inactivating ABCC8 mutation, E1506K (E1507K according to reference sequence NM_000352.2), that caused HH progression to hypoinsulinaemic DM during middle age [9, 14]. In their study, adults heterozygous for this particular mutation showed reduced insulin secretory capacity and only 1/11 adults had normal glucose tolerance [14]. In addition, the individuals who developed DM had reduced insulin sensitivity (3/4 individuals with DM were overweight). The authors hypothesised that the primary insulin secreting defect may be related to beta cell apoptosis due to raised intracellular calcium concentrations and that the accompanying insulin resistance may be the reason for conversion from impaired glucose tolerance to DM. Another family with this mutation has recently been reported with HH in childhood and later-onset diabetes [19]. Interestingly, insulin deficiency is observed in the beta cell-specific dominant-negative Kir6.2 mouse [20]. Furthermore, Abdulhadi-Atwan et al. described a patient with HH due to a de novo ABCC8 mutation who presented with DM at 10.5 years of age [12]. This child was obese (BMI 30.2 kg/m2), supporting the hypothesis that insulin resistance in conjunction with a dominant KATP channel mutation can predispose to the development of DM.
However, a recent study that looked at the prevalence of DM in a large number of families with dominant KATP channel mutations [11] reported diabetes in just 13.8% of the adult carriers. The prevalence of DM in this cohort was comparable with the estimated prevalence of type 2 DM in adults in the USA, which ranges from 9.6% to 21% [21]. In contrast we identified two adults who developed DM in adulthood following hypoglycaemia in early life and six adults who were diagnosed with either GDM or overt diabetes before the age of 50 years. Excluding the two adults who had symptoms of hypoglycaemia at the time of the study, the prevalence of DM (gestational or other) was high, with 8/14 adult mutation carriers being affected in our cohort. This may even be an underestimate as some individuals who do not have DM may go on to develop it later. In contrast, none of the 19 adult non-carriers of the ABCC8/KCNJ11 mutation within these families were known to be diabetic.
The difference in the reported prevalence of diabetes in the adult mutation carriers in our study and that of the US study [11] can be partly explained. First, there were a high percentage of adults with ongoing hypoglycaemia (51%) in the American study. The presence of hypoglycaemic symptoms in these adults signifies the persistence of hyperinsulinism. Surprisingly, these adults were not excluded from the final analysis of the adults affected with DM. Second, adults with GDM were excluded from the final reporting of patients with DM. The authors argue that the development of GDM in adults with a dominant KATP channel mutation may not have the same implication for the eventual development of diabetes as it may reflect impaired glucose-stimulated insulin release due to a direct consequence of defective KATP channel activity [11]. However, GDM per se forms an important diagnosis and has implications for the management of the individual during her pregnancy and for the growing fetus. Hence, we believe that the increased risk of GDM merits reporting in this study.
However, it is also clear that not all adult mutation carriers develop DM. We confirmed normal glucose tolerance in five of the adults who had no history of DM. In our cohort, the mean BMIs of those with normal glucose tolerance (n = 5) and those with DM (n = 8) were not significantly different. Nevertheless, the adults who developed overt diabetes following GDM in multiple pregnancies did have higher BMIs (33 and 42.2 kg/m2) than those adults who did not develop overt diabetes following GDM (23 and 26 kg/m2). However, these data must be interpreted with caution as the number of individuals in each group was small. In addition, patients with the same G1479R mutation had varied glucose tolerance, suggesting that the development of DM is not mutation specific and other environmental/genetic factors may be involved.
This study confirms that dominant ABCC8/KCNJ11 mutations can lead to a variable phenotype of neonatal HH, impaired glucose intolerance and diabetes mellitus later in life. The mechanistic explanation for the neonatal HH involves subtle defects in the function of KATP channels. We carried out Rb flux assays to prove the pathogenicity of the novel mutations identified in the study. The homozygous in vitro model was not stimulated by the KATP channel agonist diazoxide, a finding which is consistent with these mutations being disease causing. Western blotting confirmed that the mutant SUR1 proteins were produced in the cell (Fig. 2b). In patients with dominant ABCC8/KCNJ11 mutations, KATP channels are assembled from mutant and wild-type SUR1 and KIR6.2 subunits (channel hetero-octomers are formed with zero to four SUR1 and KIR6.2 mutant subunits). The eventual KATP channel activity is based on the number of mutant subunits present (in the complex consisting of both mutant and wild-type subunits) and the extent to which the mutant subunit exerts its adverse effect on channel activity. The resulting impairment in channel activity is sufficient to activate the downstream voltage-gated calcium channels and thus trigger unregulated insulin secretion. These patients respond to diazoxide presumably because some wild-type SUR1 subunits are present and allow diazoxide activation. The milder phenotype of dominant mutations reflects the subtle KATP channel defect in comparison with recessive mutations, which lead to defects in channel biogenesis, trafficking and assembly.
The mechanism of glucose intolerance and diabetes mellitus in patients with dominant ABCC8/KCNJ11 mutations is not so clear at present. Possible explanations include a progressive failure in beta cell function due to ‘exhaustion’, increased beta cell apoptosis as a result of raised intracellular calcium concentration and the influence of other genetic or environment factors. Further studies are required to understand how these mutations cause glucose intolerance and diabetes later in life.
Conclusion
Dominant mutations in ABCC8/KCNJ11 cause a varying phenotype ranging from asymptomatic macrosomia to persistent HH in childhood. There is an increased risk of DM in the adult mutation carriers. This has important implications for follow up of children with dominantly acting ABCC8/KCNJ11 mutations and the management of the adult carriers.
Abbreviations
- CHI:
-
Congenital hyperinsulinism
- DM:
-
Diabetes mellitus
- GDM:
-
Gestational diabetes mellitus
- HH:
-
Hyperinsulinaemic hypoglycaemia
- KATP channel:
-
ATP-sensitive potassium channel
- Kir6.2:
-
Inward rectifier K(+) channel Kir6.2
- SDS:
-
Standard deviation score
- SUR1:
-
Sulfonylurea receptor 1
References
Kapoor RR, James C, Hussain K (2009) Advances in the diagnosis and management of hyperinsulinemic hypoglycemia. Nat Clin Pract Endocrinol Metab 5:101–112
Thomas PM, Cote GJ, Wohllk N et al (1995) Mutations in the sulfonylurea receptor gene in familial persistent hyperinsulinemic hypoglycemia of infancy. Science 268:426–429
Thomas P, Ye Y, Lightner E (1996) Mutation of the pancreatic islet inward rectifier Kir6.2 also leads to familial persistent hyperinsulinemic hypoglycemia of infancy. Hum Mol Genet 5:1809–1812
Ashcroft FM (2005) ATP-sensitive potassium channelopathies: focus on insulin secretion. J Clin Investigation 115:2047–2058
Shyng SL, Ferrigni T, Shepard JB et al (1998) Functional analyses of novel mutations in the sulfonylurea receptor 1 associated with persistent hyperinsulinemic hypoglycemia of infancy. Diabetes 47:1145–1151
Cartier EA, Conti LR, Vandenberg CA, Shyng SL (2001) Defective trafficking and function of KATP channels caused by a sulfonylurea receptor 1 mutation associated with persistent hyperinsulinemic hypoglycemia of infancy. Proc Natl Acad Sci USA 98:2882–2887
Partridge CJ, Beech DJ, Sivaprasadarao A (2001) Identification and pharmacological correction of a membrane trafficking defect associated with a mutation in the sulfonylurea receptor causing familial hyperinsulinism. J Biol Chem 276:35947–35952
Taschenberger G, Mougey A, Shen S, Lester LB, LaFranchi S, Shyng SL (2002) Identification of a familial hyperinsulinism-causing mutation in the sulfonylurea receptor 1 that prevents normal trafficking and function of KATP channels. J Biol Chem 277:7139–7146
Huopio H, Reimann F, Ashfield R et al (2000) Dominantly inherited hyperinsulinism caused by a mutation in the sulfonylurea receptor type 1. J Clin Invest 106:897–906
Thornton PS, MacMullen C, Ganguly A et al (2003) Clinical and molecular characterization of a dominant form of congenital hyperinsulinism caused by a mutation in the high-affinity sulfonylurea receptor. Diabetes 52:2403–2410
Pinney SE, MacMullen C, Becker S et al (2008) Clinical characteristics and biochemical mechanisms of congenital hyperinsulinism associated with dominant KATP channel mutations. J Clin Invest 118:2877–2886
Abdulhadi-Atwan M, Bushman J, Tornovsky-Babaey S et al (2008) Novel de novo mutation in sulfonylurea receptor 1 presenting as hyperinsulinism in infancy followed by overt diabetes in early adolescence. Diabetes 57:1935–1940
Flanagan SE, Kapoor RR, Banerjee I et al (2011) Dominantly acting ABCC8 mutations in patients with medically unresponsive hyperinsulinaemic hypoglycaemia. Clin Genet 79:582–587. doi:10.1111/j.1399-0004.2010.01476
Huopio H, Otonkoski T, Vauhkonen I, Reimann F, Ashcroft FM, Laakso M (2003) A new subtype of autosomal dominant diabetes attributable to a mutation in the gene for sulfonylurea receptor 1. Lancet 361:301–307
Office for National Statistics (2003) Ethnic group statistics: a guide for the collection and classification of ethnicity data. www.ons.gov.uk/about-statistics/measuring-equality/equality/ethnic-group-statistics/index.html. Accessed 17 November 2010
Flanagan SE, Patch AM, Mackay DJ et al (2007) Mutations in ATP-sensitive K+ channel genes cause transient neonatal diabetes and permanent diabetes in childhood or adulthood. Diabetes 56:1930–1937
Muzyamba M, Farzaneh T, Behe P et al (2007) Complex ABCC8 DNA variations in congenital hyperinsulinism: lessons from functional studies. Clin Endocrinol (Oxf) 67:115–124
Magge SN, Shyng SL, MacMullen C et al (2004) Familial leucine-sensitive hypoglycemia of infancy due to a dominant mutation of the beta-cell sulfonylurea receptor. J Clin Endocrinol Metab 89:4450–4456
Vieira TC, Bergamin CS, Gurgel LC, Moisés RS (2010) Hyperinsulinemic hypoglycemia evolving to gestational diabetes and diabetes mellitus in a family carrying the inactivating ABCC8 E1506K mutation. Pediatr Diabetes 11:505–508
Miki T, Nagashima K, Tashiro F et al (1998) Defective insulin secretion and enhanced insulin action in KATP channel-deficient mice. Proc Natl Acad Sci USA 95:10402–10406
Signorello LB, Schlundt DG, Cohen SS et al (2007) Comparing diabetes prevalence between African Americans and Whites of similar socioeconomic status. Am J Public Health 97:2260–2267
Acknowledgements
This study was funded by the Wellcome Trust (081188/A/06/Z).
R.R.K. researched and provided data, contributed to data analysis and interpretation, and wrote the manuscript. S.E.F. researched data and contributed to the discussion and revision of the manuscript. C.T.J., A.M.T., S.C.H. and A.T. contributed to data analysis and interpretation, and draft of the manuscript. J.M.K. and J.P.S. provided data and contributed to the revision of the manuscript. S.E. and K.H. contributed to the concept, analysis of the data and revision of the manuscript. All the authors contributed to the approval of the final version of the manuscript.
Duality of interest
The authors confirm that there is no duality of interest associated with this manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Kapoor, R.R., Flanagan, S.E., James, C.T. et al. Hyperinsulinaemic hypoglycaemia and diabetes mellitus due to dominant ABCC8/KCNJ11 mutations. Diabetologia 54, 2575–2583 (2011). https://doi.org/10.1007/s00125-011-2207-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-011-2207-4